A pathway revisited
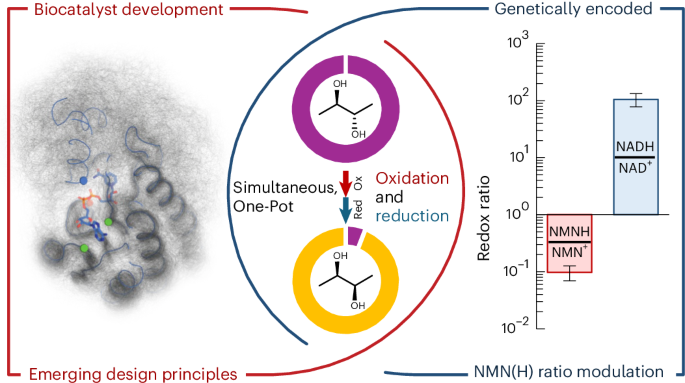
Defects in protein glycosylation and glycan metabolism lead to congenital disorders of glycosylation (CDGs). A key step in N-glycosylation involves the lipid dolichol, which in its phosphorylated form acts as a carrier of glycans to be transferred onto proteins. Previously, dolichol was thought to be directly synthesized from polyprenol by the enzyme SRD5A3. Wilson et al. have now reported a revision to this pathway, with the discovery of a three-step conversion from polyprenol to dolichol. This conversion involves an additional enzyme, DHRSX, which is also found to be associated with CDGs. The discovery first came from identifying four patients presenting with signs of CDG. Analysis of their serum glycoproteins showed aberrant glycosylation, and whole genome or exome sequencing revealed missense variants of the DHRSX gene. DHRSX was previously annotated as an NADPH-dependent oxidoreductase of unknown function. The mutations identified in the patients mapped to a hydrophobic core near a predicted NADP+ binding site in an AlphaFold2 model of DHRSX. Wilson et al. found that DHRSX produced the lipid polyprenal in the presence of polyprenol and NADP+, and SRD5A3 catalyzed the next reaction from polyprenal to dolichal. Interestingly, DHRSX also catalyzed the final reaction from dolichal to dolichol in the presence of NADPH, and both reactions involving DHRSX could occur simultaneously in vivo using NAD+ for the first reaction and NADPH for the final step. Deficiency in DHRSX led to the accumulation of polyprenol phosphates, which are poorer substrates compared to dolichol phosphates, disrupting glycosyltransferase activity. This study illustrates how the examination of four patients with CDG revealed the unique dual function of DHRSX in the metabolism of polyprenol to dolichol, critical to the initiation of the N-glycosylation pathway.
Original reference: Cell 187, 3585–3601.e22 (2024)






留言 (0)