Reagents and antibodies
Cell culture media, fetal bovine serum (FBS), antibiotics and trypsin were purchased from Life Technologies (Carlsbad, CA, USA), Immobilon-P membranes from Millipore Corp and Polyethylenimine (PEI) reagent from Polysciences (Hirschber an der Bergstrasse, Germany). Cl-amidine was purchased from Sigma-Aldrich (Sant Louis, Missouri, EEUU). Neratinib, lapatinib and tucatinib were from Selleckchem (Houston, TX, USA). Trastuzumab and T-DM1 were purchased from Farmacia Escudero, Salamanca, Spain and Farmacia Meritxell, Andorra, respectively. Other generic chemicals were from Sigma Chemicals Co, Roche Biochemicals or Merck.
Antibodies against GAPDH (sc-166574), PADI3 (#393622) and pTyr99 (sc-7020) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-calnexin antibody (SPA-860-F) from Stressgen Bioreagents (British Columbia, Canada); anti-HER2 (Ab3, #OP15) used for Western detection of human HER2 was from Calbiochem (La Jolla, CA, USA); anti-pS6 (#2215) and anti-S6 (#2217) were from Cell Signaling Technologies (Beverly, MA, USA). The anti-MGP antibody was from Proteintech (Planegg-Martinsried, Germany). The anti-FLAG antibody (F1804) was from Sigma. The horseradish peroxidase (HRP)-conjugated antibodies against mouse and rabbit IgG were obtained from GE Healthcare Life Sciences (Piscataway, NJ, USA) and Bio-Rad Laboratories (Hercules, CA, USA) and Cy3-conjugated antibodies were from Jackson Immunoresearch. Anti-human-PE was from BD Biosciences.
Cell culture, generation of clones resistant to neratinib, and cell proliferation assays
Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), containing high glucose (4500 mg/L) and antibiotics (penicillin 100 U/ml, streptomycin 100 µg/ml). Cell lines were maintained at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air. Cell authenticity was analyzed at origin by the ATCC or by STR at the Hematology Service of the University Hospital of Salamanca. Cell lines purchased from the ATCC were expanded from the initially purchased vial to ten 100 mm dishes, and when cultures were confluent, frozen 1 dish:1 vial ratio. Cells were regularly replaced (each 3-6 months) for new ones by thawing a frozen vial.
To generate clones resistant to neratinib, BT474 cells were plated in 150 mm dishes at a density of 20,000 cells per dish. Next day, once attached, cells were treated with neratinib at different doses (1 μM, 500 nM, 100 nM, 50 nM, 25 nM, 10 nM). The medium with the different neratinib concentrations was replaced once a week. After 3 months of exposure to the drug under those culture conditions, 23 clones were isolated resistant to 10 nM neratinib (numbered #1 to #23) and one clone (#24) resistant to 25 nM neratinib.
Cell proliferation was analyzed by conventional MTT uptake or cell counting experiments using a Z1 Coulter Particle Counter. Detailed protocols for these assays have previously been described [33,34,35].
Transfection, lentivirus production and infection
For transfection, PEI reagent was used. It was previously prepared by dissolving 1 mg/ml of the reagent in pre-warmed water. Once dissolved, the pH was adjusted to 7.0, filtered and aliquoted. For the transfection, 500 μl of 150 mM NaCl solution including 10 μg of DNA were mixed with 500 μl of 150 mM NaCl solution containing 40 μl of prepared PEI. After 30 min incubation, complexes were drop-wise added over the monolayers of target cells plated in 10 cm plates 24 h before. 12-24 h later media was replaced by normal culture medium before further analyses. For PADI3 overexpression, the plasmid pCMV-PADI-Myc-DKK was ordered from Origene (Herford, Germany), transfected and neomycin-resistant clones isolated (500 μg/ml, 3 weeks). To obtain a pool of SKBR3 cells overexpressing Flag-tagged PADI3, cells were transfected with the above-mentioned plasmid and selected with G418 (500 μg/ml) for a month. A pool of SKBR3 cells was analyzed for PADI3 expression by western blotting.
For lentivirus production, 4 µg of each of the plasmids pMDLg/RRE, pRSV-Rev and pMD2.G (Addgene, Cambridge, MA, USA), along with 8 µg of the pLKO.1 lentiviral plasmid containing a scramble shRNA (sh-Control) or the shRNAs for HER2 or PADI3 (GE Dharmacon, Lafayette, CO, USA) were co-transfected as described into HEK293T cells. Twenty-four hours later, HEK293T medium was replaced with fresh medium, and 48 h post co-transfection, the medium containing lentiviral particles was collected, filtered, and utilized to infect cells after the addition of 6 µg/ml polybrene (Sigma-Aldrich). Forty-eight hours after the transfection, transduced cells were selected with 3 µg/ml puromycin (Sigma-Aldrich) for another 48 h. Five distinct shRNA sequences targeting HER2 or PADI3 were assessed and used those demonstrating higher levels of knockdown (#1-ATG TAT AGG TAA CCT GTG AT and #2-TAC AAA GCC TGG ATA CTG ACA for shHER2; #36-TGG CAA GGA AGA ACA TGG TTT and #37-CCA GTA TAA GAG GGA CAA GTT for shPADI3).
Protein extraction, immunoprecipitation (IP) and Western blot (WB)
The preparation of cell extracts, IP and WB analyses were performed as described [35, 36]. Blots were visualized by autoradiography or digital capture using a Chemidoc apparatus (Bio-Rad Laboratories, Hercules, CA, USA). Densitometric measurements of the bands were performed using the Image Lab™ Software Version 6.0.1 included with the Chemidoc system.
For cell surface immunoprecipitation, cells were grown at 70-80% confluency, washed twice with Krebs-Ringer-HEPES buffer (128 mM NaCl; 5.0 mM KCl; 5.0 mM MgSO4; 1.3 mM CaCl2; 50 mM Hepes, pH 7.4) and incubated with 10 nM trastuzumab for 2 h at 4 °C in the same buffer. Monolayers were washed twice with PBS and lysed. Cell debris were removed by centrifugation, and supernatants incubated for 30 min with protein A-Sepharose. Immunocomplexes were then washed and loaded in SDS-PAGE gels as described [37].
Immunofluorescence microscopy and cell surface staining
Cells were plated on glass coverslips and 48 h later washed with PBS/Ca2 + /Mg2+ (1 mM CaCl2, 0.5 mM MgCl2 in PBS) and fixed in 2% p-formaldehyde for 30 min at room temperature. Then, cells were washed with PBS/CM, quenched with 50 mM NH4Cl and permeabilized with 0.1% Triton X-100, 0.2% BSA) and then blocked in PBS/CM with 0.2% BSA (blocking solution) for 1 h at room temperature. Monolayers were then incubated with the primary antibody in blocking solution for 2 h at room temperature. After three washes for 10 min each in PBS with 0.2% BSA, the coverslips were incubated with cyanine-3-conjugated secondary antibodies for 30 min, washed three times, 5 min each, in PBS with 0.2% BSA, and mounted. Samples were analyzed by regular epifluorescence microscopy or by confocal immunofluorescence microscopy using a Leica TCS SP5 System (Leica Microsystem CMS, Wetzlar, Germany). Phase contrast images were taken with a Nikon Eclipse Ti-S inverted microscope (Izasa, Barcelona, Spain).
To perform cell surface staining of HER2 for cytometric analyses, cells were trypsinized, collected in culture medium (DMEM + 10% FBS) and resuspended in PBS + 1% BSA. Cells were then incubated with 10 nM trastuzumab for 1 h under rocking agitation at room temperature. Later, cells were washed twice with PBS + 1% BSA and incubated with anti-human-PE (1:100) for 30 min, in agitation at room temperature. After that incubation period, cells were washed twice with PBS + 1% BSA and cytometrically analyzed using a BD AccuriTM apparatus (BD Biosciences). Mean fluorescence intensity of HER2 was quantified using the BD Accuri C6 Software.
Microarray transcriptomic studies
Total RNA extraction was performed with the RNeasy Mini kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. RNA concentration was determined with a NanoDropTM 1000 apparatus (Thermo Fisher Scientific). Assessment of the quality of the isolated RNAs was carried out by the Genomics Service of our Institute, using the Bioanalyzer RNA 6000 Nano assay (Agilent). cDNAs were hybridized to Clariom S human arrays (Affymetrix, Santa Clara, USA). Raw CEL files were normalized using the RMA algorithm and the genes that presented differential expression were identified using the Transcriptome Analysis Console (TAC) software version 4.0.2.15 (Affymetrix). The cut-off values taken were fold-change <-2 or >2 and a p-value of ANOVA < 0.01.
HER2 sequencing
Total RNA was isolated as described above and first-strand cDNA was synthesized using M-MLV reverse transcriptase and oligo-dT (Invitrogen, Carlsbad, CA, USA), following the instructions from the manufacturer. The oligonucleotides used to amplify HER2 are described in supplementary fig. 2B. The PCR reactions were as follows: 5 min at 94 °C and 35 cycles of: 30 s at 94 °C, 30 s at 56 °C, 1 min at 72 °C. Next 7 min at 72 °C. After PCR, the amplified fragments were separated on agarose gels and the bands were visualized in a Gel Doc 2000 (Bio-Rad Laboratories, Hercules, CA, USA) apparatus. Bands were excised and amplified DNAs purified using the GFXTM PCR DNA and gel Band purification KIT (GE Healthcare, Buckinghamshire, United Kingdom). DNA concentration was measured in a NanoDropTM 1000 apparatus, and 100 ng used for sequencing. The sequences obtained were compared with the canonical HER2 sequence, available in the databases (Ensembl) (transcript ID ENST00000269571.10), using the Clustal Omega web tool.
Statistics
Data distributions were checked for normality by the Shapiro-Wilk test and homogeneity of variances was checked by the Levene test. The Mann–Whitney test was used to compare continuous variables between two groups and Anova for >2 groups. Statistical analyses were performed using GraphPad Prism 8 (San Diego, CA, USA).

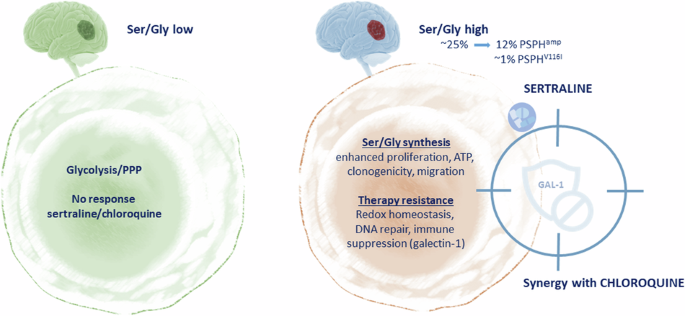







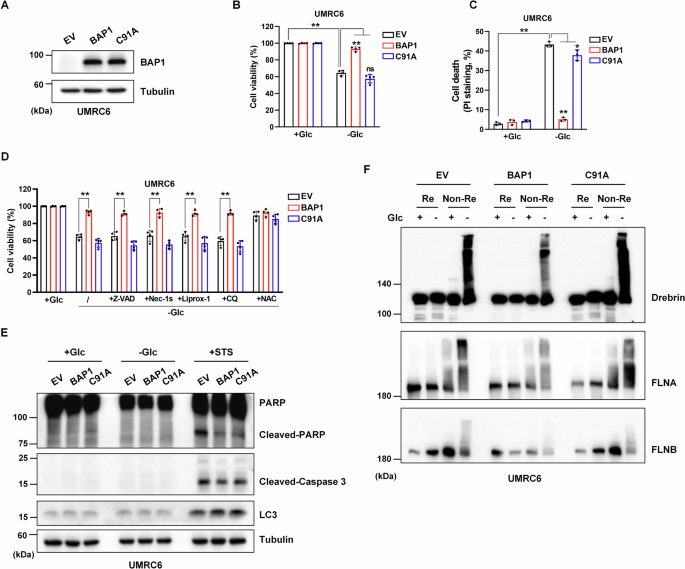
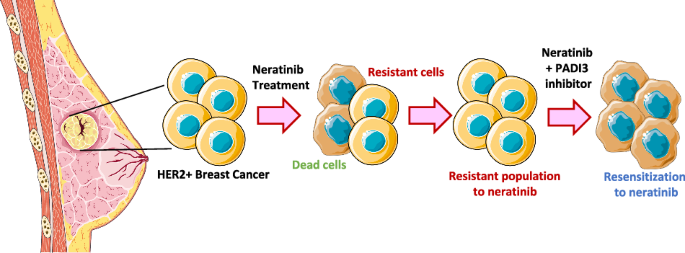
留言 (0)