
記住我
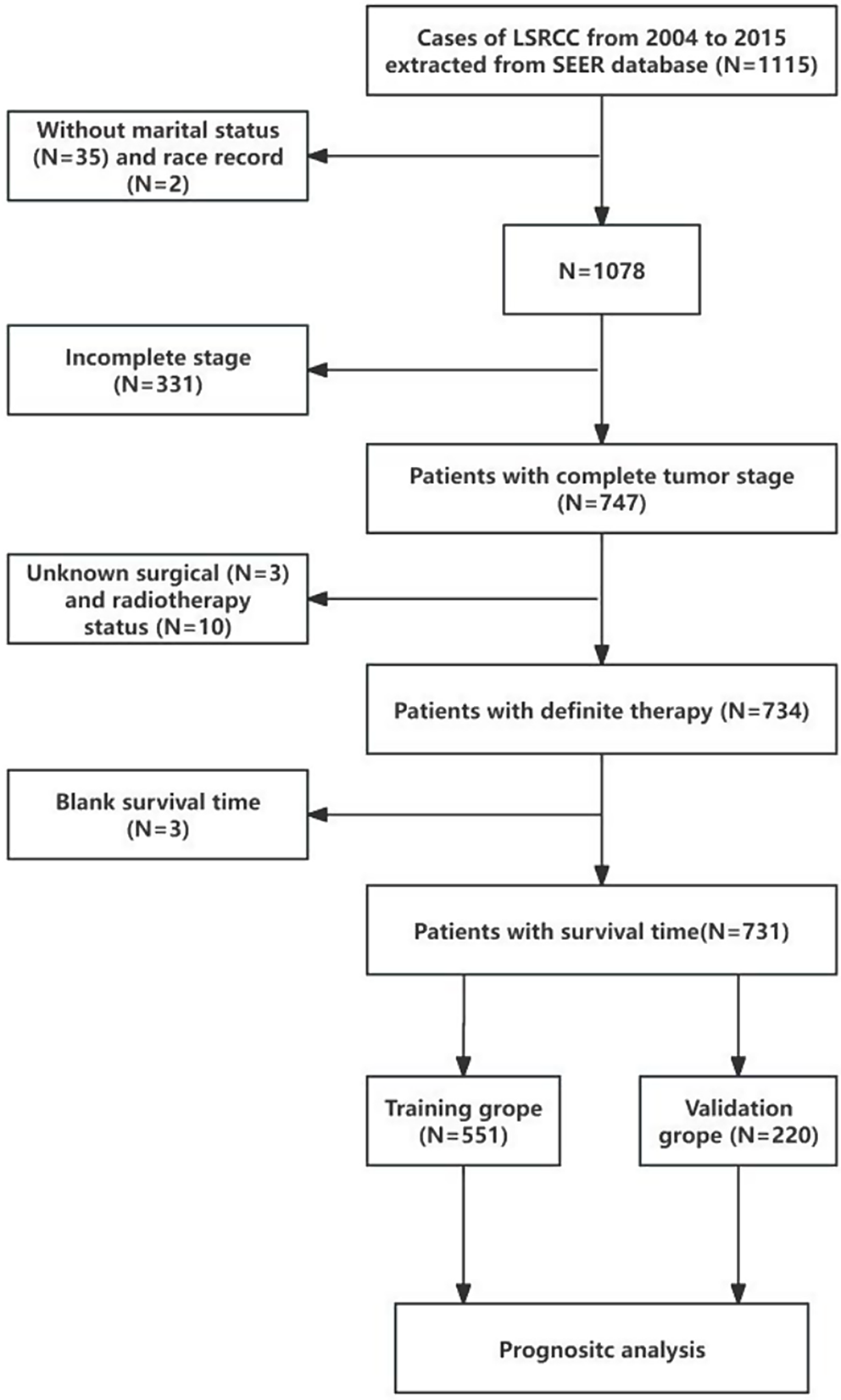





A comprehensive analysis revealed 21,186 DEGs between PCa and normal groups, consisting of 12,229 up-regulated and 8,957 down-regulated genes. Eight DE-CRGs (DLAT, SLC31A1, PDHA1, CDKN2A, DLD, GLS, ATP7B, FDX1) were highlighted, integrating data from 13 CRGs and DEGs (Fig. 1A-B). These eight DE-CRGs are distributed across five chromosomes and exhibit notable correlations among them. Notably, DLD exhibited a significant positive correlation with DLAT, while SLC31A1 showed a marked negative correlation with PDHA1 and CDKN2A (Supplementary Fig. 1).
Fig. 1
Identification of Cuprotosis-related differentially expressed genes in prostate cancer. (A) Heat map of Cuprotosis-related genes in prostate cancer patients. The colors in the heatmap signify the correlation, with red representing a positive correlation and blue representing a negative correlation. (B) Box plot of Cuprotosis-related genes in prostate cancer patients
Gene expressions of the mentioned 8 DE-CRGs were utilized to identify two clusters among 338 patients with survival data in the TCGA-PRAD dataset (cluster 1 = 238, cluster 2 = 100) through NMF analysis (Fig. 2A, Supplementary Table 1). Most genes exhibited consistent expression trends within the same clusters (Fig. 2B). Survival analysis revealed that patients in cluster 2 had a poorer prognosis compared to cluster 1 (Fig. 2C). Consequently, cluster 2 was chosen as the clinical trait for subsequent WGCNA analysis.
Fig. 2
Identification of Cuprotosis clusters in prostate cancer patients. (A) NMF analysis results. The k value at which the resonance correlation coefficient starts to decrease was chosen and the optimal number of clusters was 2. (B) Expression heatmap of Cuprotosis-related genes in two types of prostate cancer patients. Blue represents the first type and yellow the second. The higher/lower the level of expression, the darker/lighter the colour. (C) Survival curve plots of prostate cancer patients in two clusters
Identification of 6561 cluster 2-related genes and 322 Gleason score-related genesAfter excluding two outliers via sample clustering, the WGCNA analysis included 336 samples with associated DFI information from the TCGA-PRAD dataset (Supplementary Fig. 1). Selecting a soft threshold power of 6 ensured the establishment of a scale-free network (R^2 = 0.85), as shown in Fig. 3A. Subsequently, 6 co-expression modules were generated based on similar gene expression patterns (Fig. 3B-C). Among these modules, MEturquoise displayed the most significant negative correlation with cluster 2 (|Cor| = -0.35, p.adj < 0.0001) (Fig. 3D). Consequently, a total of 6561 genes within the MEturquoise module were identified as key module genes (referred to as cluster 2-related genes) for downstream analysis. The correlation between GS and MM is depicted in Fig. 3E, demonstrating the robust association between these module genes and cluster 2, indicating a poorer prognosis.
Fig. 3
Identification of 6561 Cuprotosis clustering related genes in prostate cancer patients. (A) Determination of the WGCNA soft threshold. (B-C) WGCNA generates co-expression modules. (D) Heatmap of the correlation between 6 co-expression modules and Cluster 2. The values in the heatmap represent the correlation, and the values in the parentheses represent the p-value. Red indicates positive correlation, while blue indicates negative correlation. (E) Detection of the correlation between module genes and prognosis in prostate cancer patients
Sample clustering was conducted on 497 samples from the TCGA-PRAD dataset, excluding two outliers (Supplementary Fig. 3). A soft threshold power of 9 (Fig. 4A) was set to establish a scale-free network (R^2 = 0.85), leading to the identification of 14 co-expression modules (Fig. 4B-C). The heatmap illustrating Module-Gleason relationships revealed that MEmagenta displayed the strongest association with Gleason scores (|Cor| = 0.39, p.adj < 0.0001) (Fig. 4D). Consequently, a total of 322 genes within MEmagenta were discovered as genes related to Gleason score. The correlation between GS and MM of these genes is depicted in Fig. 4E.
Fig. 4
Identification of 22 Gleason score-related genes. (A) Determination of the WGCNA soft threshold. (B-C) Generation of co-expression modules by WGCNA. (D) Screening of modules related to Gleason score in prostate cancer patients. The values in the heatmap indicate the correlation, and the values in parentheses denote the p-values, with red indicating a positive correlation and blue indicating a negative correlation. (E) Detection of the correlation between module genes and Gleason score in prostate cancer patients
Identification and exploration of 27 candidate genesIntegrating Gleason score-related genes and cluster 2-related genes identified a total of 27 candidate genes (Fig. 5A, Supplementary Table 2). GO enrichment analysis revealed their enrichment primarily in various biological process (BP) terms, such as DNA biosynthetic process, centriole replication, and centriole assembly. Cellular components (CC) included chaperonin-containing T-complex and chaperone complex, while molecular functions (MF) encompassed single-stranded DNA helicase activity, suggesting potential associations with these genes (Fig. 5B, Supplementary Table 3). In terms of potential KEGG signaling pathways, five terms were implicated, including DNA replication and Cell cycle (Fig. 5C, Supplementary Table 4).
Fig. 5
Identification and exploration of 27 candidate genes. (A) VENN diagram of Gleason score and genes related to copper mortality. (B) GO enrichment analysis of candidate genes. The x-axis shows the ratio number of genes and the y-axis shows the GO pathway terms. The size of the dots indicates the number of enriched genes in the pathway, and the colour represents the range of p-values. (C) KEGG enrichment analysis of candidate genes. The x-axis shows the ratio number of genes and the y-axis shows the KEGG pathway terms. The P-value of each term is colored according to the legend
Construction and validation of the prognostic risk modelTo investigate prognostic implications in PCa, we selected five out of the 27 candidate genes (STX3, CABLES2, E2F5, RALA, POLE3) utilizing univariate Cox and LASSO Cox regression analyses (p < 0.05) to build a prognostic risk model (Fig. 6A-B, Supplementary Table 5). Risk scores were computed for 338 patients in the training set based on the gene expression of these five prognostic genes, leading to patient stratification into high- and low-risk groups. Principal component analysis (PCA) plot revealed distinct discrimination between the two risk populations (Fig. 6C), consistent with the risk curves (Fig. 6D-E). Additionally, a significant correlation was observed between shorter DFI and higher risk scores according to Kaplan-Meier analysis (p < 0.0013) (Fig. 6F). Time-dependent ROC curves demonstrated that the area under the curve (AUC) for DFI at 1-, 3-, and 5-years were 0.81, 0.79, and 0.72, respectively (Fig. 6G).
Fig. 6
Construction of prognostic risk model. (A-B) Univariate Cox and LASSO Cox regression analysis to construct the prognostic risk model. (C) PCA cluster analysis. (D-E) Risk curves of the training set. (F) K-M survival curve analysis of patients in the training set. (G) ROC curve for predicting 1–5 year survival of patients in the training set
Likewise, we computed the risk score for each patient in the validation set (GSE70769) (Fig. 7A-B). When correlated with BRF data, it became apparent that patients in the high-risk groups demonstrated a worse prognosis (p = 0.0057) (Fig. 7C), with AUC values surpassing 0.6, confirming the predictive accuracy of the prognostic risk model (Fig. 7D).
Fig. 7
Validation of the prognostic risk model.(A-B) Risk curves in the validation set. (C) K-M survival curve analysis in the validation set patients. (D) ROC curve for predicting 1–5 year survival in the validation set patients
The prognostic risk model was associated with the muscle system process and cardiac muscle contraction pathwayWe investigated the underlying mechanism associated with the prognostic risk model. A total of 165 DEGs were identified between the high- and low-risk groups (|Log2FC| > 1.5, p.adj < 0.05) (Fig. 8A-B), consisting of 44 up-regulated genes and 121 down-regulated genes. Enrichment analysis revealed that the GO processes primarily involved activities related to the muscle system process, contractile fiber, and sarcomere (Fig. 8C, Supplementary Table 6). Conversely, only 5 KEGG pathways were implicated, including cardiac muscle contraction and hypertrophic cardiomyopathy (Fig. 8D, Supplementary Table 7).
Fig. 8
Prognostic risk model related to muscle system processes and myocardial contraction pathway. (A) Volcano plot of differential genes between high-risk group and low-risk group. (B) Heatmap of differential genes between high-risk group and low-risk group. The colors in the heatmap signify the correlation, with red representing a positive correlation and blue representing a negative correlation. (C) GO enrichment analyses were performed on 165 DEGs, listing the top 30 GO entries. (D) KEGG enrichment analysis of 165 DEGs
Patients with high expression of STX3 have larger tumor volumes later staging higher Gleason scores and worse prognosisIn the research, we identified STX3 as a significant predictor for the prognosis of PCa patients, an aspect underexplored in PCa research. We collected specimens from clinical PCa patients, with their basic clinical information summarized in Table 1. Immunohistochemical staining was performed on tissues obtained from all patients (Fig. 9A). The results revealed significantly higher expression levels of STX3 in PCa patients in comparison with healthy individuals, with statistical significance (Fig. 9B). According to STX3 expression levels, patients were classified into high expression (n = 46) and low expression (n = 47) groups. Interestingly, STX3 expression was significantly elevated in PCa patients with low differentiation, stage III + IV, and lymph node metastasis compared to those with high differentiation, stage I + II, and absence of lymph node metastasis (Table 1). Additionally, patients with high STX3 expression exhibited larger tumor volumes and higher Gleason scores (Table 1). However, STX3 expression did not significantly correlate with other clinical pathological features (Table 1). The 5-year survival rates of patients with high and low STX3 expression were 49.29% and 68.17%, respectively. Moreover, the total survival period of patients with low STX3 expression was notably higher than that of patients with high STX3 expression (Fig. 9C).
Table 1 Comparison of STX3 expression levels in PCa patients with different pathological features(n(%))Fig. 9
Differential expression of STX3 in PCa patients. (A) Immunohistochemical staining of prostate cancer group and normal control group patients. (B) Statistical analysis of immunohistochemical staining in prostate cancer group and normal control group patients. (C) Survival curve of PCa patients with different STX3 expression levels
STX3 promotes the proliferation, migration, and invasion of prostate cancer cellsTo explore STX3’s influence on prostate cancer cell behaviors like proliferation, migration, and invasion, we established PCa cell lines with STX3 knockdown. In the PC-3 cell line, STX3 mRNA expression notably decreased (independent samples t-test, P < 0.01), as shown in Fig. 10A. Western blot analysis further confirmed a significant reduction in STX3 expression in the sh-STX3 group compared to the sh-NC group (Fig. 10B), validating the efficacy of STX3 knockdown.
Fig. 10
STX3 inhibits proliferation migration and invasion of prostate cancer cells in vitro. (A) Relative expression levels of STX3 mRNA in the sh-STX3 group and sh-NC group. (B) Protein expression levels of STX3 in the sh-STX3 group and sh-NC group. (C) OD450 values of CCK8 experiments in sh-STX3 group and sh-NC group. (D) Colony formation results in sh-STX3 group and sh-NC group. (E) Wound healing rate at 24 h in the sh-STX3 group and sh-NC group. (F) Number of migrated cells in the sh-STX3 group and sh-NC group. (G) The number of invading cells in the sh-STX3 group and sh-NC group
CCK-8 assays conducted 24 h post-transfection exhibited a notably reduced proliferation rate within the sh-STX3 group in comparison with the sh-NC group in the PC-3 cell line (Fig. 10C). Similarly, clonogenic assays demonstrated a significant decrease in clonogenic potential within the sh-STX3 group in comparison with the sh-NC group (Fig. 10D). These findings collectively suggest that downregulation of STX3 suppresses the proliferation of PCa cells.
Migration assays (Fig. 10E-F) demonstrated a significant reduction in the migration rate of the sh-STX3 group compared to the sh-NC group in the PC-3 cell line. This suggests that downregulating STX3 expression suppresses the migration ability of PCa cells. Additionally, Transwell invasion assays conducted in the PC-3 cell line revealed a significantly decreased number of cells passing through the Transwell chamber in the sh-STX3 group compared to the sh-NC group (Fig. 10G), indicating that STX3 downregulation attenuates the invasion capability of PCa cells.
STX3 promotes the proliferation of prostate cancer cells in miceUsing the PC-3 cell line, we established subcutaneous tumor models in nude mice. Upon dissection, it was evident that the volume of subcutaneous tumors in the sh-STX3 group was significantly smaller than that in the sh-NC group (Fig. 11A). Quantitative analysis of tumor volume from the 10th day post subcutaneous tumor cell implantation showed a significant reduction in tumor volume in the sh-STX3 group compared to the sh-NC group (Fig. 11B). Furthermore, the weight of each tumor tissue post-dissection revealed a substantially lighter tumor weight in the sh-STX3 group compared to the sh-NC group (Fig. 11C). Immunohistochemical staining confirmed STX3 expression in tumor tissues (Fig. 11D).
Fig. 11
Suppression of STX3 Expression Reduces Prostate Cancer Cell Growth and Tumor Burden in Nude Mouse Models. (A) Tumor size in sh-STX3 group and sh-NC group mice. (B) Tumor volume size in sh-STX3 group and sh-NC group mice. (C) Difference in tumor weight between sh-STX3 group and sh-NC group mice. (D) Immunohistochemical staining of STX3 mouse tumor and its statistical results
In conclusion, suppressing STX3 expression led to diminished PCa cell growth in nude mice, resulting in decreased tumor volume and burden on the animals.
留言 (0)