
記住我
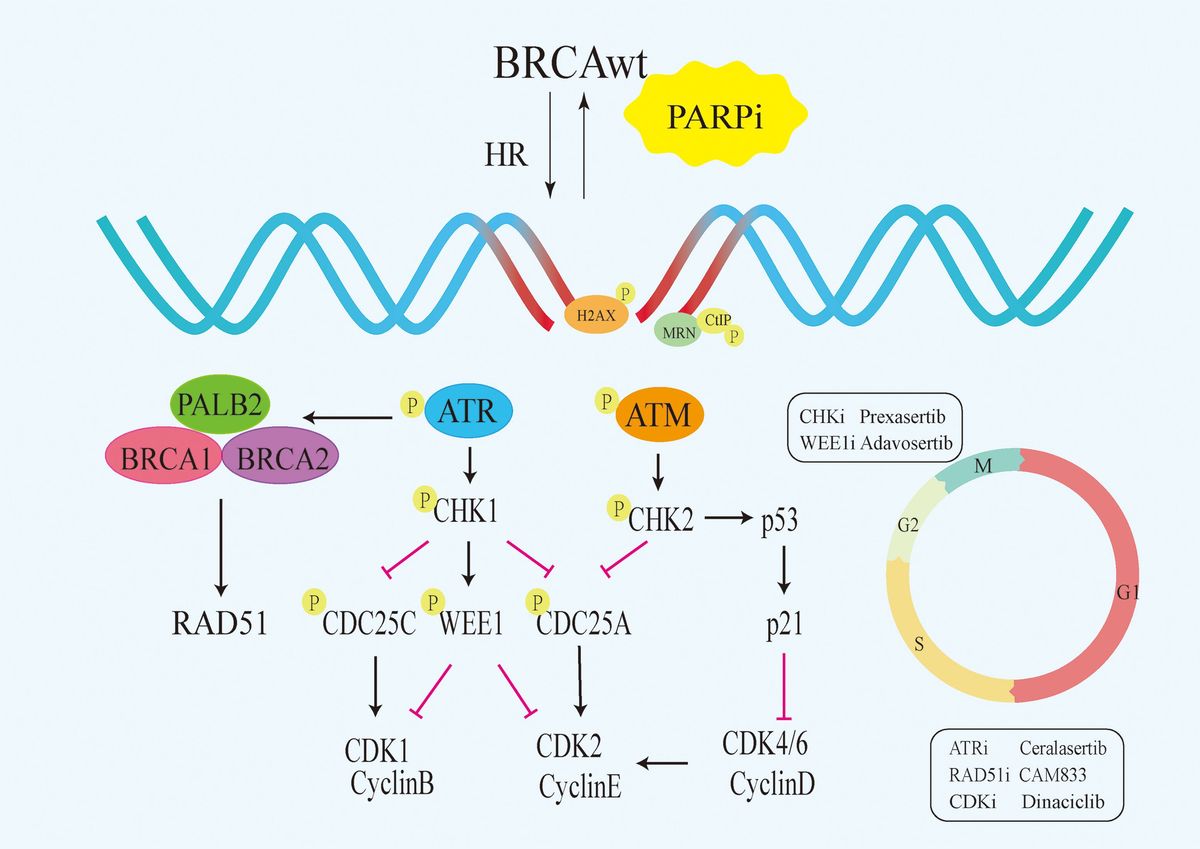
Base excision repair (BER) is the primary mechanism for removing oxidized bases producing single strand break (SSB) intermediates. PARP1 (and PARP2) recognizes SSB and gaps in the DNA and catalyzes the addition of chains of ADP-ribose molecules to proteins in a process known as poly-ADP-ribosylation (PARylation) (1). PAR facilitates BER/SSBR by recruiting DNA repair factors and chromatin remodellers. PARP1/2 not only plays a crucial role in the repair of SSB (2). When PARP1/2 is deleted or inhibited, SSB accumulates and encounters replication forks during proliferation, which converts to DNA double-strand damage (DSB) (3). DSB is the most toxic DNA damage in cells, which is mainly repaired by high-fidelity HR pathway (4). When HR is deficient, DSB are primarily repaired through error-prone non-homologous end joining (NHEJ) pathway, and erroneous DNA repair can lead to genomic instability and cell death (5). Therefore, simultaneously inhibiting PARP1/2 and HR will lead cells to undergo synthetic lethality. Synthetic lethality refers to the phenomenon that the simultaneous inactivation of two genes will lead to cell or individual death, while the cell or individual can survive normally when either gene is inactivated alone. The application of the concept was first validated clinically in cancers with BRCA1 and BRCA2 mutations leading to HR repair deficiency (6, 7). Subsequently, PARP inhibitors (PARPi) have developed rapidly in clinical trials for ovarian and breast cancers with BRCA1/2 mutations and other HR gene defects. Currently, six oral clinical PARPi effectively inhibit the catalytic function of PARP1 and PARP2 by competing with NAD+ for binding to PARP1/2 (8). Olaparib, Rucaparib, Niraparib, and Talazoparib have been approved by the Food and Drug Administration (FDA) for ovarian, breast, pancreatic and prostate cancer patients with BRCA mutations (8). In addition to catalytic inhibition, most PARPi also possess the ability to trap PARP on DNA, thereby preventing PARP1 protein from undergoing PARylation modification and releasing from damaged sites (9, 10). The formation of PARP1-DNA complex, which is trapped on the DNA, hinders the progression of replication fork and promotes DSB formation (10), which is the major mechanism by which PARPi kills HR-deficient cancer cells (10, 11). PARPi have high trapping capacity and can effectively kill cells with HR defects.
Although PARPi have achieved great success in the treatment of BRCA mutated patients, it has limited efficacy in BRCA1/2 wild-type patients. According to research data, only about 20% of high-grade serous ovarian cancer patients who have BRCA mutations are more sensitive to PARPi, while the remaining approximately 80% of BRCA wild-type patients do not benefit from PARPi (12–14). Similarly, approximately 80% of triple-negative breast cancer (TNBC) patients without BRCA mutations do not respond to PARPi (15, 16). Due to the intact HR pathways in BRCA wild-type cells, even if PARPi prevent the repair of SSB and subsequent occurrence of DSB, tumor cells can still maintain chromosome stability and cell viability through HR (17). Consequently, the exploration of utilizing PARP inhibitors for treating BRCA1/2 wild-type cancer patients has piqued the interest of researchers. Studies have revealed that, besides BRCA1/2, proteins such as MRN, ATM, CHK1, CtIP, RAD51 play direct roles in the HR process (Figure 1). Upon DNA damage, the MRE11A-NBS1-RAD50 (MRN), serves as a damage sensor by detecting DNA damage and binding to the break end (18). This complex then mediates the activation of ataxia telangiectasia mutated (ATM), which leads ATM to switch from an inactive dimer to an active monomer, phosphorylates sites at Ser-367, Ser-1893, Ser-1981, and Ser-2996 (19). These signals are relayed to CHK1 and BRCA1, while CtIP is ubiquitinated to facilitate S and G2 arrest (20, 21). Subsequently, RAD51 binds to the damaged DNA ends, forming DNA-RAD51 nucleoprotein filaments. At the same time, RAD51 can recognize a sister strand that matches the damaged DNA sequence, enabling recombination exchange between the two strands for DNA repair (22). Considering this, a basic strategy is to combine PARPi with drugs that may induce HR defects. For example, inhibiting ATR, CHK, and RAD51 to downregulate HR can enhance the sensitivity of BRCA wild-type patients to PARPi (23). Furthermore, the combination of PARP inhibitors with drugs that indirectly interfere with HR, such as VEGFR, BRD4, EZH2, HDAC, and PD-L1 inhibitors, results in synthetic lethality in BRCA1/2 wild-type tumors, expanding the scope of PARP inhibitor applications (24–26) (Figure 2). Apart from HR inhibition, synergistic effects can be achieved by combining chemotherapy drugs or radiotherapy with PARP inhibitors, as those leads to further enhancement of DNA damage. In this review, we summarize the basic principles of combining PARP inhibitors in wild-type BRCA cancers, ongoing clinical trials, and analyze the future directions of PARPi combination therapy.
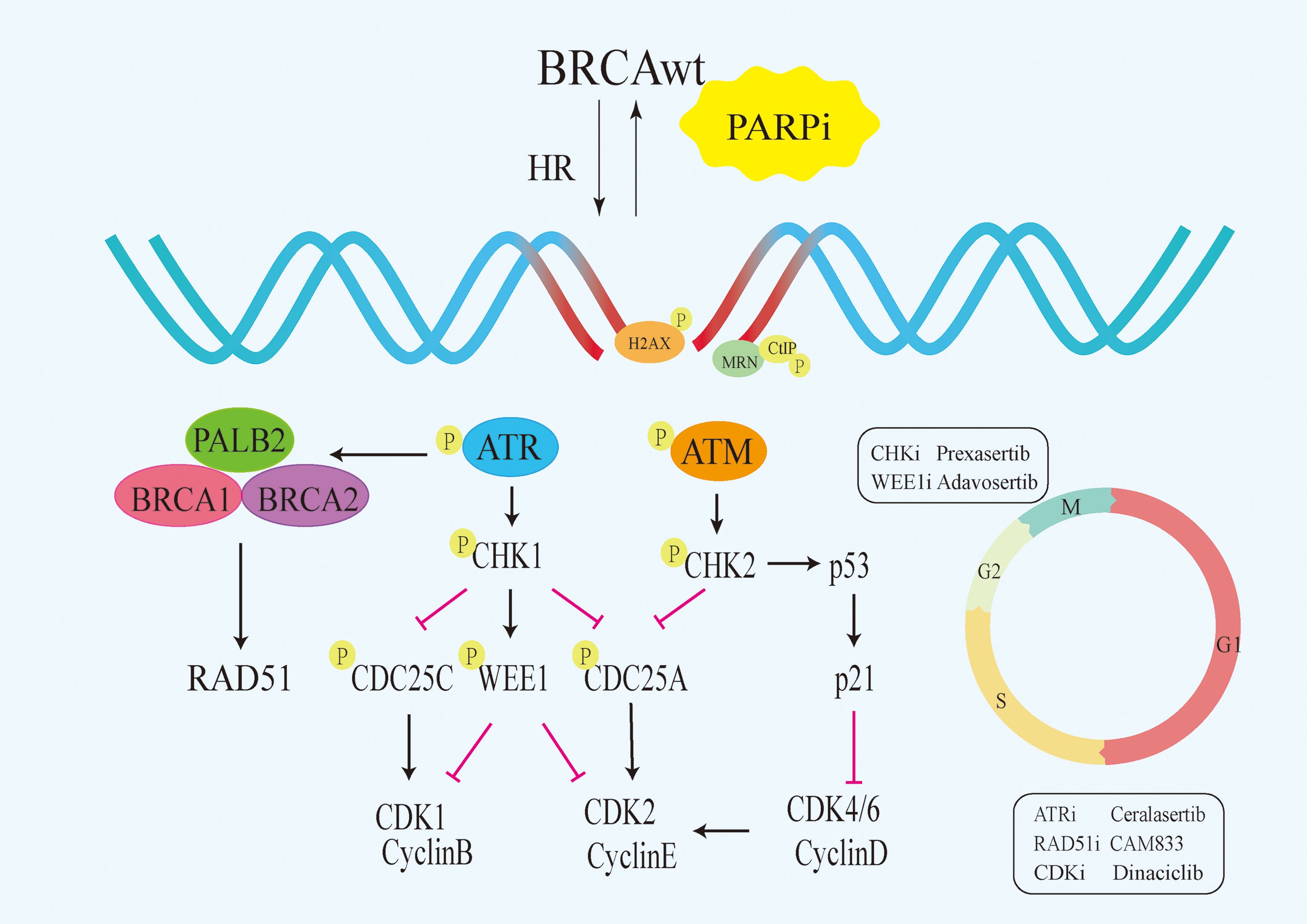
Figure 1 Signalling pathways that directly target HR. PARPi,PARP inhibitors;HR, homologous recombination;BRCAwt, BRCA wild-type;ATM, Ataxia Telangiectasia Mutated.
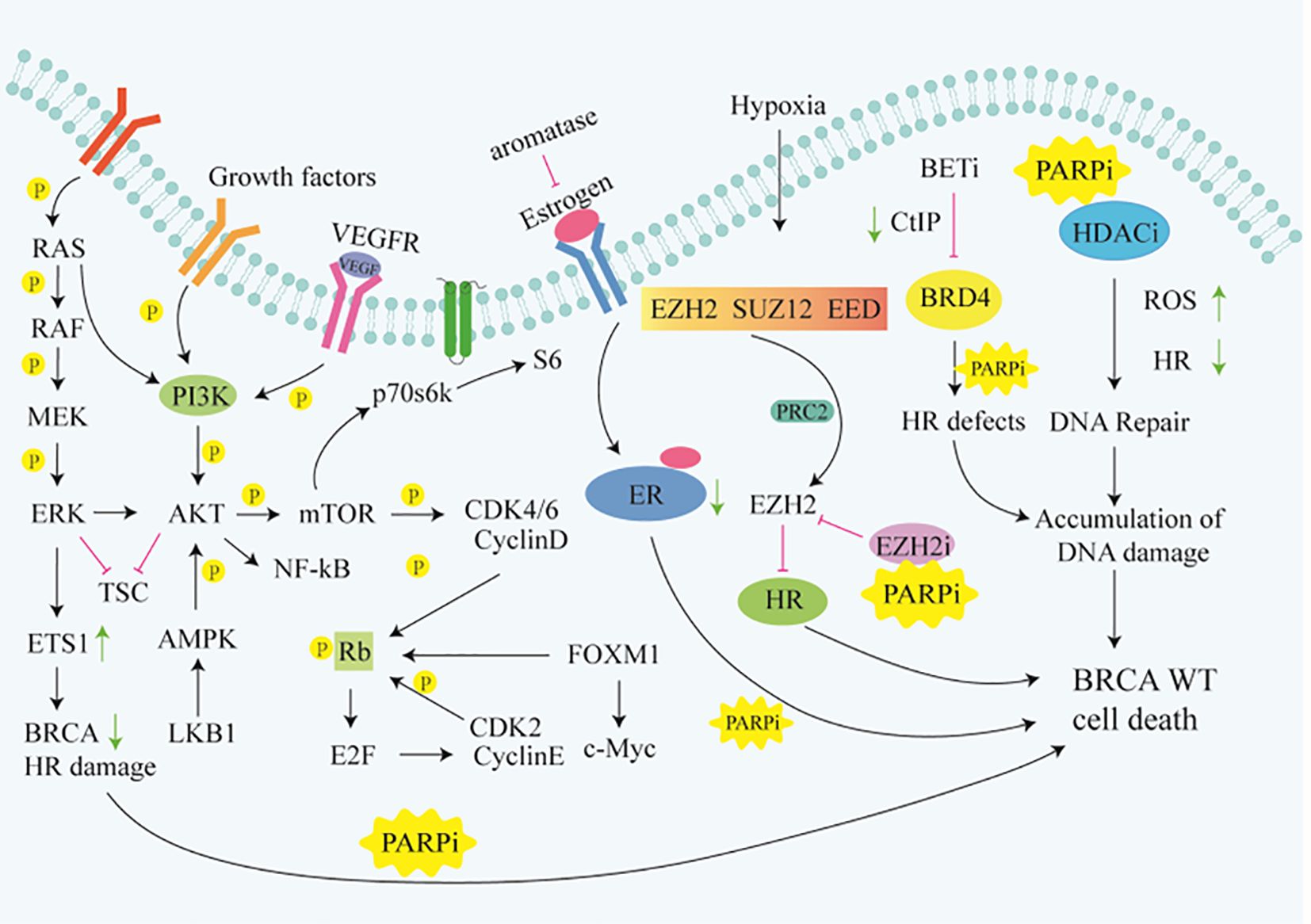
Figure 2 Signaling pathways that indirectly target HR. Rb, Retinoblastoma gene;ER, Retinoblastoma gene;TSC, Tuberous Sclerosis Complex; FOXM1,Forkhead box M1.
2 Inhibitors that directly suppress HR2.1 ATR inhibitorsATR (ataxia telangiectasia and Rad3-related) is a member of the Phosphoinositide 3-kinase (PIKK) family of serine/threonine protein kinases (27). ATR is a key regulatory factor of the DNA damage response (DDR) pathway, working together with other DDR proteins to initiate and coordinate the cell’s response to DNA damage and stress (28). Activated ATR kinase regulates various cellular processes, including inducing cell cycle arrest, replication initiation inhibition or restart and DSB repair (29–33). Multiple studies have identified BRCA1 as a target of ATR. BRCA1 expression is regulated by E2F, which in turn can be controlled by the ATR-CHK1 pathway (34–36). Prolonged chronic inhibition of ATR signaling depletes abundance of key HR factors such as BRCA1 and PALB2, significantly suppressing the cell’s ability to utilize HR-mediated DNA repair. Long-term treatment with ATR inhibitors(ATRi) can render HR-proficient cancer cells sensitive to PARPi (37). ATR promotes HR by phosphorylating PALB2 and enhances its repair of DNA damage by interacting with BRCA1 (38). Inhibition of ATR kinase aims to target tumor cells highly sensitive to high replication stress. While inhibiting ATR activity may induce replication fork stalling and collapse in normal cells, leading to some cytotoxicity, this cytotoxicity is further exacerbated in cancer cells with high replication stress (39–41). PARPi alone are insufficient to kill BRCA wild-type cancer cells, but their combination with ATRi shows synergistic inhibitory effects on cancer cells, leading to increased DNA damage (42). ATRi in combination with PARPi synergistically induce tumor cell death through inhibiting DNA repair pathways, leading to a synthetic lethality effect (43–45). Phosphorylation of Histone H2AX at Ser-139 is a marker of DNA damage. The ATR inhibitor Ceralasertib (AZD6738) can block CHK1 phosphorylation and increase γH2AX expression (46, 47). Preclinical studies have shown significant synergistic efficacy of Ceralasertib in combination with Olaparib in BRCA wild-type triple-negative breast cancer (TNBC) xenograft models, achieving complete tumor regression by increasing the dosage of Olaparib or Ceralasertib to twice daily (48).
Many patients develop resistance to PARPi, with acquired PARPi resistance being a major obstacle in treating tumors. A study on a patient-derived xenograft (PDX) model for platinum-resistant BRCA wild-type patients showed that the combination of ATRi and PARPi have a synergistic effect, leading to increased DNA damage and sustained regression of ovarian tumors, significantly improving patient survival (49). Currently, there is no definitive data on clinical trials of PARPi-ATRi therapy for BRCA wild-type patients. Additionally, identifying cancer types sensitive to combination treatments and optimizing combination dosages are key to the successful clinical application of combined PARPi and ATRi therapy in BRCA1/2 wild-type cancer patients, necessitating further large-scale studies.
2.2 CHK inhibitorsResearch indicates that DNA damage repair enables cells to respond to various stresses threatening genome stability. This response involves two main signaling pathways, ATM/Chk2 and ATR/CHK1, with the dysfunction of CHK1/2 closely linked to tumorigenesis (50, 51). CHK1 is a major effector downstream of ATR, becoming phosphorylated at Ser317 and Ser345 sites by the ATR kinase during replication stress-induced DNA damage (52, 53). While CHK2 acts as a serine/threonine kinase, it is a downstream phosphorylation substrate of ATM and is activated when DSB occur (54, 55). Further mechanistic studies showed that ATR-mediated phosphorylation and activation of CHK1 induced cell cycle arrest after DNA damage, which in turn led to the phosphorylation and degradation of cell division cycle factor 25 (CDC25), thereby inhibiting cell cycle progression to mitosis (M) and ultimately leading to cell cycle arrest to gain time for DNA repair. In contrast, when CHK1 is inhibited, CDC25 will be dephosphorylated, leading to the activation of C8/CDK complex. Although the presence of DNA damage will lead to programmed cell death, this complex will still drive cells to complete cell cycle progression. The primary functions of CHK1 not only involve regulating the cell cycle to prevent premature entry into the M phase but also stabilizing stalled replication forks and regulating HR (56, 57). Overall, CHK inhibitors (CHKi) play a crucial role in promoting cancer cells apoptosis through the aforementioned pathways.
In the process of homologous recombination repair after DNA damage, the binding of RAD51 to BRCA2 depends on the CHK1 phosphorylation of the C-terminal domain of BRCA2. RAD51 is responsible for regulating the invasion of ssDNA into the complementary parental strand to generate an extendable primer-dsDNA. CHK1-mediated phosphorylation of BRCA2 is important for the stability of BRCA2 and the efficient recruitment of RAD51 to sites of DNA damage, thereby facilitating effective DNA repair through homologous recombination (58, 59). CHK1 repairs excessive DNA damage in cancer cells, while inhibition of CHK1 leads to downregulation of DNA repair protein RAD51 and enhanced DNA damage (60). Therefore, CHK1i block the activation of HR by inhibiting the localization of RAD51 to nuclear lesions during DNA damage, thereby inhibiting the function of RAD51. The above studies elucidate the important role of CHK1i in blocking cell cycle progression, weakening replication forks, and inhibiting HR. DDR is coordinated by CHK1 and CHK2, acting independently to delay cell cycle progression and provide time for DNA repair. The ability of CHK1 to inhibit HR enhances the efficacy of PARPi on BRCA proficient tumors (61). Additionally, PARPi treatment upregulates p-ATR and p-CHK1, indicating the activation of the ATR-CHK1 protective pathway is critical in PARPi resistance. In fact, combination therapy with ATRi or CHK1i and PARPi synergistically reduces the survival and colony formation of BRCA wild-type cancer cells compared to monotherapy. Notably, PARPi treatment leads to cell cycle G2 arrest, while ATRi or CHK1i induces premature entry into mitosis, increasing chromosomal aberrations and cancer cell apoptosis (62). The combination of PARPi and CHK1i increases DSB and γH2AX expression. Studies have shown that ATM knockdown inhibits drug-induced CHK1 and ERK1/2 phosphorylation and enhances the cytotoxicity of PARPi and CHK1i on tumor cells (63) Prexasertib (LY2603618) is the first selective CHK1/2 inhibitor. Hye-Yon Cho found that the combination therapy of Prexasertib with Rucaparib exhibits significant anticancer effects in BRCA wild-type ovarian cancer cell lines, consistent with the study by Hyoung Kim (64). Based on these preclinical studies, the potential of CHK1i and PARPi combination therapy as a novel treatment for BRCA wild-type patients have been confirmed.
2.3 RAD51 inhibitorHistone H2AX and RAD51 are key proteins involved in the DNA repair pathway (65, 66). Histone H2AX is phosphorylated into γH2AX, leading to the formation of other DNA repair proteins, such as RAD51, at the site of DSB (67, 68). Consequently, in preclinical and clinical samples, the formation of γH2AX and RAD51 foci are used as a biomarker for DSB (69). RAD51 and its family play multiple roles in DSB repair, replication stress, and meiosis. Downregulation of RAD51 expression reduces the DNA damage repair capacity of tumor cells, thereby enhancing the efficacy of tumor gene toxic therapy (70). It has been reported that RAD51 is a key protein that mediates HR, and RAD51 chromatin loading is the core step of HR (71). As a downstream effector molecule of the BRCA2 protein, RAD51 can be loaded onto ssDNA to promote the formation of RAD51-ssDNA nucleoprotein filaments and catalyze strand exchange reactions to initiate homology-directed repair (HDR) (72, 73). In fact, RAD51 foci are known biomarkers for the HR repair pathway in vitro. When tumor cells are exposed to DNA-damaging agents, RAD51 is recruited to the sites of DNA damage and forms distinct foci in a proficient HR repair environment (74). Targeting the specific interaction of RAD51-BRCA2 can mimic the effect of BRCA deficiency. Data suggest that in BRCA1 wild-type TNBC, strategies targeting RAD51 can enhance the therapeutic efficacy of PARP inhibitors (75). Additionally, Scott et al. found that RAD51 inhibitor CAM833 enhances the damage effects of PARP inhibitors on BRCA wild-type cells by blocking the BRCA2-RAD51 protein interaction and preventing RAD51-mediated HR (76). In conclusion, RAD51 is the core protein of HR, and the combination of its inhibitor and PARPi is a promising treatment for BRCA wild-type tumors, which deserves further study.
2.4 WEE1 inhibitorsWEE1 is a type of tyrosine kinase, functioning as a key regulatory factor of the G2-M cell cycle checkpoint (77). The expression of WEE1 in cancer cells has dual biological roles. As a tumor suppressor, WEE1 can delay cell entry into mitosis by inhibiting CDK activity, thereby maintaining genomic stability. However, loss of WEE1 may lead to the accumulation of genetic abnormalities, promoting the development of pre-neoplastic lesions. As an oncogene, WEE1 promotes cancer cells to evade the effects of DNA damage and abnormal mitosis (78). WEE1 as an oncogene is highly expressed in various cancer types, including breast cancer. Cancer cells often exhibit defects in the G1-S checkpoint and heavily rely on the G2/M checkpoint to resist endogenous and exogenous DNA damage (79, 80). Activated WEE1 during DNA damage response maintains ATR and CHK1 phosphorylation to delay cell entry into mitosis (81). Similar to ATR, WEE1 is also involved in replication fork protection through direct interaction and negative regulation of DNA cleavage by endonuclease MUS81 (82). The MUS81 has a structure-specific activity for the Holliday junction formed during HR (83). Inhibition of WEE1 activates CDK1, leading to phosphorylation of BRCA2 and slowing down the progression of replication forks, thereby limiting HR (84–86). Since Adavosertib (AZD1775) usually regulates the G2/M checkpoint, the combination of Olaparib and AZD1775 significantly attenuates G2 arrest. AZD1775 reduces the expression of CtIP and RAD51 and disrupts HR repair (87). The increased susceptibility of cells to DNA damage induced by PARPi is due to the lack of CDK1/2-mediated phosphorylation-induced DNA repair defects and WEE1 inhibition(WEE1i).
Research has shown that combination therapy using PARPi and WEE1i synergistically induces cell death through replication stress and DNA damage (88). Cyclin E, as a key cell cycle regulatory factor, is a biomarker mediating replication stress in cancer. In tumor cells, it accumulates in the cytoplasm in a low molecular weight form. By co-administering Niraparib and WEE1i (Adavosertib) acting on Cyclin E, apoptosis in BRCA wild-type cells can be accelerated (89). Studies by Teo et al. have also demonstrated the synergistic effect of combining these two drugs in controlling tumor growth. The combination of Olaparib and Adavosertib triggers an increased anti-tumor immune response, including activation of the STING pathway. Combined use with STING (stimulator of interferon genes) agonists can further enhance persistent tumor regression in BRCA1/2 wild-type TNBC mouse tumor models, significantly improving survival outcomes (90).
2.5 CDK inhibitorsThe progression of the cell cycle largely depends on cyclin-dependent kinase (CDK), and the imbalance of CDK is associated with two key features of cancer cells, cell cycle dysregulation, and abnormal proliferation (91). The CDK-RB-E2F axis constitutes a central transcriptional mechanism that drives cell cycle progression, dictates the timing and fidelity of genome replication, and ensures the accurate transmission of genetic material in each cell division cycle (92). E2Fs are the main transcriptional regulatory factors of cell cycle-dependent gene expression, and they are highly active in almost all cancers, usually due to the inactivation of their main binding partner and key regulator RB (retinoblastoma), overexpression of CDK, or inactivation of CDK inhibitors (CDKi) (93, 94). Studies have confirmed that cyclin and its catalytic part control the transition between different stages of the cell cycle (95). Cyclin D1 is considered a key oncogenic driver in cancer (96), promoting the G1/S phase transition by binding and activating CDK4 and CDK6 (97). CDK4/6i effectively block cancer cell proliferation by inducing G1 cell cycle arrest (98). Furthermore, Cyclin D1 can inhibit Cyclin A-CDK2-dependent Ser329 phosphorylation and promote the binding of RAD51 to the C-terminal domain of BRCA2, while down-regulation of Cyclin D1 leads to low HR efficiency (99). Interfering with the HR repair pathway by blocking CDK1 can mimic BRCA1 mutations and increase the sensitivity of TNBC cells to PARPi by 100-fold (100). Wild-type BRCA also participates in G1 cell cycle arrest. Aprelikova found that BRCA1 binds to hypophosphorylated RB and interacts with the E2F transcription factor to block transcription and inhibit cell proliferation (101). Research has shown that inhibiting CDK1, suppressed and transformed BRCA1 expression and phosphorylation transforms BRCA wild-type cancer cells into HR-deficient cells, making them more susceptible to synthetic lethality induced by PARPi (102, 103).
Additionally, the Johnson et al. discovered that the CDKi Dinaciclib reduces the expression of HR genes in BRCA wild-type TNBC cells and sensitizes these cells to Veliparib. Another study developed a dual PARP and CDK6 inhibitor named P4i, which is a new compound that links PARPi Olaparib and CDK6i Palbociclib through an o-phenylenedione moiety. This inhibitor significantly induces DNA damage and cell apoptosis, inhibiting the proliferation of TNBC cells through the signaling pathways involving PARP1 and CDK6 in BRCA wild-type cells (104). However, the interplay between the Cyclin/CDK pathway, HR, and BRCA wild-type remains complex, requiring further exploration and confirmation through future randomized clinical trials.
3 Inhibitors that indirectly suppress HR3.1 VEGFR inhibitorsThe vascular endothelial growth factor (VEGF or VEGF-A) was initially identified as a vascular permeability factor (VPF) and is one of the key molecules associated with angiogenesis (105). Patel pointed out that the angiogenic pathway, which develops new blood vessels from the existing vascular system, is a crucial step in tumor growth and metastasis (106). Studies have shown that hypoxia-induced by anti-angiogenic therapy inhibits HR repair by suppressing the expression of key factors such as BRCA1 and BRCA2, leading to a deficiency in DDR and rendering BRCA1/2 wild-type cancer cells sensitive to PARPi (107, 108). BRCA wild-type tumor cells are more sensitive to the VEGFR3 inhibitor Maz51. The addition of Maz51 can lead to BRCA gene down-regulation, inducing cell cycle arrest and leading to BRCAness, benefitting BRCA wild-type patients treated with PARPi (109). Furthermore, numerous studies have reported a synergistic effect of VEGFR inhibitors combined with PARPi in reducing the proliferation and invasion capabilities of tumor cells (110–112).
Early preclinical studies have shown that the anti-angiogenic agent Cediranib can inhibit the pro-survival and anti-apoptotic AKT signaling, significantly enhancing the inhibitory effects of ribonucleotide reductase inhibitor Triapine and Olaparib on BRCA wild-type epithelial ovarian cancer cells and extending the survival time of mice (113). Results from the clinical study NT02354131 also indicate that the combination of Bevacizumab and Niraparib is more effective than Niraparib monotherapy in treating BRCA wild-type ovarian cancer (114). Similarly, another phase III study showed that the combination of Bevacizumab and Olaparib than placebo plus bevacizumab significantly prolonged progression-free survival (PFS) in patients without BRCA gene mutation (28. 1 months vs 16. 6 months) (115). In addition, a phase II study also showed that the combination of Cediranib and Olaparib significantly improved the overall survival of patients with BRCA1/2 wild-type ovarian cancer (37. 8 months vs 23. 0 months) (116). Subsequently, another phase III trial of J. F Liu showed that the remission rate of patients in the Olaparib/Cediranib combination group was significantly improved (117). Through preclinical and clinical studies, the efficacy of combining PARPi with VEGFR inhibitors have been widely recognized. Li et al. developed the first dual VEGFR/PARP inhibitor, which inhibits angiogenesis and invasion by negatively regulating the expression of VEGFR and PARP, thereby suppressing the growth and metastasis of BRCA wild-type breast cancer (118). These studies highlight the advantages of VEGFR/PARP dual inhibition in treating BRCA wild-type patients, with the potential to benefit more patients in the future. However, the exact mechanisms of combination therapy are not fully understood and may vary depending on the specific VEGFR inhibitors used, thus further research is needed to elucidate the precise mechanisms through which these combination therapies exert their anti-cancer effects.
3.2 EZH2 inhibitorsEpigenetic modifications are closely associated with tumorigenesis, mainly through regulating gene function and expression levels via DNA methylation and Histone modifications, thereby controlling cell differentiation (119). Enhancer of zeste homolog 2(EZH2) is the catalytic subunit of polycomb repressive complex 2(mainly composed of EZH2, embryonic ectoderm development (EED), suppressor of zeste 12 (SUZ12)), also a major regulator of cell cycle progression, autophagy, and apoptosis (120), and its overexpression can promote DNA damage repair and tumorigenesis (121). Therefore, any dysregulation of EZH2 might facilitate cancer development, while inhibiting its function and expression could make it an ideal target for epigenetic drug therapy. Numerous studies suggest that the combination of EZH2 inhibitors(EZH2i) and PARPi exhibits improved anticancer activity (122, 123). The typical mechanism involves tri-methylation of H3K27me3 by catalyzing EZH2, mediating transcriptional silencing to inhibit HR, thereby synergistically inducing synthetic lethality with PARPi (122, 124–128). Given the role of EZH2 as a transcriptional regulator, extensive research efforts have been devoted to identifying downstream targets or pathways driven by EZH2. In addition, mounting evidence indicates that EZH2 also plays a non-canonical role as a transcriptional activator, activating oncogenic pathways in a PRC2-independent manner, and directly modulating the activity of transcription factors and other proteins (129, 130). For instance, EZH2 transcriptionally upregulates IDH2 and promotes ovarian cancer growth by enhancing the tricarboxylic acid cycle activity to facilitate OXPHOS (131). However, the combination of EZH2i and PARPi can also elicit negative effects in the tumor microenvironment (TME). For example, the dual loss caused by PARP1 and EZH2 due to PRC2 deficiency exerts an oncogenic effect in BRCA wild-type breast cancer, primarily activating the NF-κB signaling pathway by forming a ternary complex with RelA and RelB (121), inducing the differentiation of tumor-promoting M2-type macrophages, disrupting the TME (132). Nevertheless, whether EZH2 inhibitors can fully suppress its biological functions to the extent of EZH2 loss remains inadequately studied.
GSK126 is one of the earliest discovered two selective EZH2i. It is more than 1000 times selective to EZH2 compared to other 20 methyltransferases, capable of reducing H3K27me3 levels and reactivating silenced PRC2 target genes (133). By 2020, the latest generation of EZH2i Tazemetostat, also known as a competitive inhibitor of SAM, was approved by the FDA (134). Tazemetostat may have higher selectivity, better pharmacokinetic properties, better clinical efficacy and fewer side effects compared to GSK126, these advantages make it a more potential EZH2 inhibitor. In 2021, Wang et al. designed the first PARP and EZH2 dual inhibitor for treating BRCA wild-type TNBC, and its anti-proliferative activity was 15-80 times higher than that of Olaparib in BRCA wild-type cells (135), suggesting that the combination of EZH2i and PARPi holds promising prospects in the future.
3.3 HDAC inhibitorsAs anti-tumor drugs, HDAC inhibitors(HDACi) can regulate the expression of HR-related genes, induce cell cycle arrest, apoptosis, and differentiation in tumor cells, leading to oxidative stress and DNA damage (136, 137). Preclinical studies have shown that Histone H3-Ser10 is a major target for ADP-ribosylation, and specific Histone acetylation marks have been found to block this activity. Among them, ADP-ribosylation induced by DNA damage is inhibited by jointly destroying post-translational modification (PTM) pathways such as acetylation-ADP-ribosylation (138). It has been reported that the HDACi SAHA induces acetylation of Histone H3 and induces degradation of the UHRF1 protein which is involved in maintenance DNA methylation and DNA damage repair. Combination therapy with Veliparib and SAHA can synergistically reduce the levels of BRCA1 by targeting the UHRF1/BRCA1 protein complex, and decreased UHRF1 levels can lead to BRCA1 protein degradation (139). New evidence indicates that dysregulation of HDAC function leads to downregulation of DNA repair genes such as RAD51, BRCA1/2, causing DNA repair defects and accumulation of DNA damage. Inhibition of HDAC can enhance the anti-tumor effect of PARPi in TNBC patients by blocking the DNA repair pathway (140).
Research has shown that the combination therapy of PARPi and HDACi significantly enhances the sensitivity of BRCA wild-type tumor cells to PARPi (141, 142). Synergistic effects of PARPi and HDACi have been observed in various cancer cells in vitro and in vivo studies. HDACi can inhibit DNA damage repair, downregulate HR and induce “BRCAness”, enhancing the biological activity of PARPi in TNBC regardless of BRCA1 mutation status (143, 144). Researchers have found that the anti-tumor efficacy of HDACi is partially attributed to the downregulation of PARylation, inhibiting DNA repair proteins. This repair inhibition, combined with cancer cell-specific high levels of reactive oxygen species (ROS) and DNA replication stress, makes cancer cells highly sensitive to HDACi/PARPi combination therapy (145). As early as 2017, Yuan et al. constructed hydroxamic acid derivatives of Olaparib as dual inhibitors of PARP and HDAC, which significantly induced apoptosis in MDA-MB-231 cells (146). In conclusion, the combination therapy of PARPi and HDACi holds great promise as a cancer treatment, providing strong theoretical support for the treatment of BRCA wild-type patients, with more clinical trials to validate these research findings in the future.
3.4 BRD4 inhibitorsBromodomain-containing protein 4 (BRD4) belongs to the bromodomain and extra-terminal domain (BET) family of proteins. Acting as a key factor in chromatin structure and transcription regulation, BRD4 plays a significant role in DNA damage repair and cell proliferation (147, 148). The survival of cancer cells is known to rely on aberrant transcription driven by super-enhancers (SEs), providing valuable targets for cancer treatment. Inhibiting BRD4 can disrupt the communication between SEs and promoters, effectively suppressing the transcription and expression of oncogenes, reducing cancer cell proliferation and viability. Specific inhibition of oncogenes leads to tumor cell death, making this mechanism the most recognized action of BET inhibitors(BETi) (149, 150). Further research has shown that inhibiting BRD4 can downregulate the transcription levels of 7 MTC(m6A methyltransferase complex) components, resulting in an overall decrease in m6A(N6-methyladenosine) modification. BETi significantly downregulate multiple genes in the HR pathway through the MTC-m6A mechanism, while also upregulating several pro-apoptotic genes. Studies using PDX models have revealed the synergistic effect of BETi/PARPi in targeting tumors through the BRD4-MTC-HR signaling axis (151). Recent studies indicate that regardless of BRCA1/2 status, inhibiting BRD4 with BETi decreases the expression of the DNA repair factor CtIP, inducing HR defects and enhancing DNA damage induced by PARP inhibitors in cancer cells (152). These findings not only confirm the synergistic effects of BETi with DDR-targeted drugs but also demonstrate the potential of extending the efficacy of PARP inhibitors to non-BRCA1/2 mutant cancers.
Yang et al. ‘s study demonstrates that inhibiting or depleting BET proteins affects the transcription of BRCA1 and makes various tumor cells sensitive to PARP inhibitors (153). JQ1, as the first extensively studied BET family inhibitor, competitively binds to the acetylated lysine recognition motif or bromodomain (154). Preclinical studies indicate that the combination treatment of JQ1 with Olaparib can reduce the IC50 of Olaparib in OVCAR3 cells by approximately 50-fold, synergistically inhibiting the growth of BRCA1/2 wild-type cells through inducing apoptosis (155). Further research reveals that simultaneous damage to the HR and BER pathways can induce significant death in BRCA wild-type TNBC cells. This implies that BET directly regulate HR-mediated DNA repair and induce the BRCAness phenotype in BRCA1 wild-type TNBC cells (156). However, since many BRD4 inhibitors are pan-BET inhibitors rather than solely targeting BRD4, there is a risk of off-target effects. Therefore, the development of PARP/BRD4 dual inhibitors have become one of the future research directions. Wang et al. synthesized a highly selective PARP/BRD4 dual inhibitor, which displayed good synergistic anti-tumor efficacy in BRCA wild-type PDAC cells by blocking the cell cycle progression, inhibiting DNA damage repair, and promoting autophagy-related cell death (157). In 2022, another researcher designed a dual inhibitor (BP44) that can block G0/G1 transition and cell mitosis, reverse Olaparib-induced adaptive resistance, inhibit DNA damage repair, and promote DNA damage to induce death in BRCA wild-type TNBC cells (158). These findings collectively suggest that the combination of PARPi and agents specifically targeting BRD4 may present a novel strategy and direction for treating patients with BRCA wild-type cancers.
3.5 PI3K inhibitorsThe dysregulation or mutation of the PI3K/AKT/mTOR pathway is one of the most common aberrant activation pathways in human malignancies, with increased PI3K signaling also considered as a hallmark of cancer (159, 160). Inhibiting the PI3K signaling pathway induces feedback upregulation of ERK, leading to increased activation of the ERK-related transcription factor ETS1 (161, 162). As ETS1 is a negative regulator of BRCA1/2 expression, the upregulation of ETS1 results in downregulation of BRCA and HR damage, making BRCA wild-type TNBC cells sensitive to PARP inhibitors (163–165). Studies have shown that treatment with Olaparib and Rucaparib leads to significant upregulation of the PI3K/mTOR pathway (p-mTOR, p-AKT, and pS6). Additionally, negative regulators of the PI3K pathway such as LKB1 and its targets AMPK and TSC are significantly downregulated. Following PARP depletion, phosphorylation of mTOR, AKT, and S6 increases while LKB1 signaling diminishes (166). Preclinical studies indicate that the combination of the PI3K inhibitor Buparlisib (NVP-BKM120) and Olaparib delays tumor proliferation in mouse models for over 70 days and in BRCA1-related tumor xenograft models for over 50 days, suggesting that combined PI3K and PARP inhibition could be an effective treatment for BRCA1-related tumors (167). BKM120 blocks PI3K by reducing HR ability, increasing ROS-mediated accumulation of γH2AX and DNA oxidative damage and inhibiting the expression of BRAC1/2 and RAD51/52 (168). BKM120 downregulates the expression of PARP1 and PARP2 to assist in PARP-mediated SSB repair blocking by Olaparib through the PI3K/Akt/NFκB/c-Myc signaling pathway and PI3K/Akt/FOXM1/Exo1 signaling pathway inhibiting HR. The combination of PI3K inhibitor BKM120 and Olaparib significantly reduces the proliferation of BRCA-proficient TNBC cells (169).
Based on successful preclinical studies, several clinical trials have demonstrated that the combination of PI3K/PARP inhibitors sensitizes BRCA wild-type TNBC, ovarian, and breast cancers to PARPi (170, 171). In 2020, researchers synthesized the first PARP/PI3K dual inhibitor, which can significantly inhibit the growth of BRCA wild-type cells by inhibiting the PI3K signaling pathway, down-regulating BRCA expression and inducing DNA damage and apoptosis (172). At present, an increasing number of dual inhibitors targeting BRCA wild-type cells have been developed, demonstrating superior anti-proliferative characteristics (173, 174).
3.6 Estrogen receptorsEstrogens regulate cell growth and development by acting on two different estrogen receptors ERα and ERβ. Among them, ERα can drive up to 70% of breast cancer, therefore targeting estrogen-positive receptors (ER+) is the standard approach for treating metastatic breast cancer (175, 176). Additionally, as a steroid hormone, estrogen plays a crucial role in maintaining sexual and overall health by regulating gene expression through interactions with transcription factor proteins in the cell nucleus rather than directly binding to DNA (177, 178). Since the BRCA1 gene is a susceptibility gene for breast cancer, it can regulate the proliferation and differentiation of breast cells (179). During puberty and pregnancy, estrogen levels rise rapidly, leading to excessive mammary gland development and promoting the expression of BRCA1 wild-type gene (180). Early studies indicate that by modulating the transcriptional activity of ERα to limit its stimulatory effect on proliferation of mammary epithelial cells, the occurrence of breast cancer can be effectively suppressed (181, 182). Furthermore, BRCA1 wild-type can also reduce estrogen levels by inhibiting aromatase expression, thereby further decreasing ERα-mediated transcriptional activity (183).
As mentioned earlier, estrogen can inhibit cancer cells through various pathways. Recent research has shown that estrogen can suppress the expression of the BRCA1 gene by stimulating the release of nitric oxide in ER+ breast cancer cells. This results in the accumulation of DSBs based on H2AX foci formation, reducing the HR repair mechanism in wild-type BRCA cancer cells, thereby enhancing the sensitivity of BRCA wild-type tumors to PARPi (184). On the other hand, as the most important and active hormone in estrogen, estradiol (E2) is closely related to the proliferation, differentiation and DNA damage of breast cells. Preclinical trials have shown that the combination of PARPi and E2 has a synergistic effect, with E2 enhancing PARPi-induced DNA damage and effectively inhibiting the recurrence of BRCA1/2 wild-type tumors (185). Endocrine therapy for breast cancer was one of the earliest molecular targeted therapies used in cancer treatment, and besides causing endocrine-related symptoms, it does not lead to severe adverse events. The combination of PARPi and targeted estrogen therapy in breast cancer treatment has significant advantages and may become one of the important strategies in the field of personalized medicine in the future.
4 The combined application of PARP inhibitors and immune checkpoint inhibitorsPD-1 is a cell surface molecule that regulates adaptive immune responses. It transmits signals inhibiting T cell proliferation, cytokine production, and cytotoxic function by binding to its ligands, PD-L1 or PD-L2 (186, 187). High expression of PD-L1 serves as a prognostic biomarker for tumor progression and predicts the efficacy of immune checkpoint inhibitors (ICIs) in certain cancers (188). In recent years, multiple studies have also demonstrated an association between tumor immunity and HR, providing a theoretical basis for the combined use of PARPi and ICI (189–191). For instance, PD-L1 can promote DNA end resection to regulate HR in BRCA1 wild-type tumor cells, enhancing HR repair capacity in tumor cells. Thus, the lack of PD-L1 can lead to increased DNA damage accumulation and improved tumor control of PARP inhibition in BRCA1 wild-type tumors, while triggering synthetic lethality to PARP inhibitors in vitro (192).
On the other hand, PARPi can also indirectly activate dendritic cells by activating the cGAS-STING signaling pathway (193). PARPi increases the number of dendritic cells and enhances the antigen presentation mechanism by inducing the upregulation of two different signals, the co-stimulatory proteins CD80 and CD86, and MHC II (major histocompatibility complex class II), which mediates the presentation of antigens to T cells (194, 195). These two signals are also key components in the activation of naïve T cells, which after activation can control the expression of cell surface receptors such as CTLA-4 and PD-1 (196). PARPi-mediated DDR rapidly increases type I interferon expression through the cGAS-STING pathway and is a favorable factor in the treatment of ICI (197). DNA damage and cytoplasmic DNA-mediated cGAS- STING pathway contributes to the remodeling of the immune supportive environment. In addition, a series of studies have shown that PARPi-mediated DNA damage can enhance T cell recruitment and infiltration by activating the cGAS-STING pathway (194, 198, 199). For example, niraparib can activate STING-mediated type I interferon release and enhance T cell infiltration in tumors. In BRCA wild-type tumor models, inducing changes in the tumor microenvironment favored its combination with ICIs showing synergistic anti-tumor activity (200). Thus, combining PARPi to target these co-inhibitory pathways of ICIs in the context of cancer could be effective in achieving long-term anti-tumor effects. A mechanistic rationale for the use of PARPi as an immunomodulator to harness the therapeutic benefits of immunotherapy is provided.
The combination of ICI and PARPi has shown a synergistic effect in preclinical studies involving BRCA wild-type cancer, leading to the advancement of multiple clinical trials. A phase II clinical study of Olaparib and Durvalumab in the combination therapy for recurrent ovarian cancer has demonstrated that the combination of PARPi and anti-PD-L1 creates an immune stimulatory environment that can enhance durable anti-tumor immune response in the BRCA wild-type population toward immune checkpoint blockade (201). In the phase II trial (NCT03167619), the PFS of Olaparib in combination with Durvalumab was significantly longer compared to historical controls, with the subgroup of platinum-sensitive advanced TNBC patients with BRCA wild-type gaining sustained disease control (202). Another phase I and II clinical trials of Niraparib in combination with Pembrolizumab have shown an improved efficacy in tBRCA wild-type compared to monotherapy (ORR, 19%). Notably, among 8 patients with a response duration of over 6 months, 5 had platinum-refractory or platinum-resistant ovarian cancer and tBRCA wild-type tumors (203). Furthermore, the use of MEK inhibitors/PARPi combination or MEK inhibitors/PARPi/anti-PD-L1 triple therapy in BRCA wild-type tumor patients in NCT03695380 has demonstrated overall ORR and PFS superiority over single-agent Rucaparib (204, 205). The triple therapy combining ICI, PARPi, and Bevacizumab in the MEDIOLA trial (NCT02734004) has also shown promising activity, especially in BRCA wild-type tumor patients (206). The combination of ICI with PARPi may be a potential approach to enhance PARPi anti-tumor activity; however, these efficacy results still need to be confirmed in phase III clinical trials and compared to controls using PARPi alone.
5 The combined application of PARP inhibitors and chemotherapyChemotherapy, as a widely recognized cancer treatment method, typically uses cytotoxic drugs to treat various cancers (207). Chemotherapeutic agents are generally designed to kill tumor cells and prevent their proliferation, thereby inhibiting further growth and spread of the tumor. Chemotherapy can also cause DNA damage through various mechanisms, contributing adjunctively to enhancing the efficacy of other treatments, such as targeted therapies (208). For example, alkylating agents are the most common chemotherapy drugs, acting to chemically modify DNA at the level of base pairs. This DNA base damage may not immediately induce cytotoxic effects but rather induce cell cycle arrest by disrupting replication forks, leading to further cell damage like replication-associated DSB, mitotic catastrophe and cellular apoptosis. Considering the critical role of DSB repair pathways in cancer therapy resistance, inhibitors targeting different DSB repair pathways have been developed as potential sensitizers for conventional cancer treatments (209). Among them, PARPi play a pivotal role in blocking the DSB repair mechanism, which can maintain DSB damage and eventually lead to tumor cell death. Therefore, combination chemotherapy is more commonly used than single therapy.
Despite the strong theoretical basis for the combination therapy of PARPi and chemotherapy, it has not been proven to be as effective as other targeted HR inhibitors treatment strategies so far. One major issue is the narrow therapeutic window of drug treatment, as the synergistic effect of chemotherapy combined with PARPi is non-selective for tumor cells. Particularly, when PARPi is combined with platinum-based drugs, it enhances chemotherapy toxicity, including hematologic toxicity (210). A Phase III VELIA trial (NCT02470585) demonstrated no difference in ORR and PFS in the combination group compared to the control group in the BRCA wild-type population (211). Furthermore, clinical trials with paclitaxel regimens showed that weekly dosing improved PFS in BRCA wild-type patients compared to every three weeks dosing (carboplatin and paclitaxel with Veliparib) (18. 0 months vs 12. 9 months). However, while increasing the dosage to enhance efficacy, toxicity also increased accordingly (212). A Phase II clinical trial (NCT02595905) showed that adding Veliparib to cisplatin improved progression-free survival in BRCA1/2 wild-type metastatic triple-negative breast cancer patients compared to cisplatin with placebo (18. 3% vs 4. 7%). Although the study showed that the Veliparib combination improved PFS, the exact conclusion on the efficacy of the combination therapy stage could not be drawn due to the potential impact of previous ICI treatment on Veliparib treatment effectiveness (213). The combination of chemotherapy and PARPi leads to additive toxicities such as bone marrow suppression, limiting patient treatment. Optimizing combination therapy regimens (including dosage and administration sequence) to reduce side effects while maintaining efficacy is a challenge that needs to be addressed when combining PARPi targeting HR repair with chemotherapy drugs.
6 The combined application of PARP inhibitors and radiotherapyRadiotherapy (RT) is one of the cornerstones of cancer treatment, involving the use of high-energy ionizing radiation (IR) to kill cancer cells (214). The ideal treatment scenario involves selectively damaging only the tumor cells throughout the body while minimizing damage to healthy cells (215). However, the clustered DNA damage caused by IR does not always result in cancer cell death but rather triggers various complex DNA repair processes such as BER (216, 217). In such cases, PARPi are used as radiosensitizers to enhance the effects of radiation on tumors, improving anti-tumor responses with lower toxicity (218). The combination with ionizing radiation can enhance the radiosensitivity of various tumor cells, driving tumor cell death (219–224). The mechanism involves PARPi blocking the repair of radiation-induced damaged DNA through the BER pathway, increasing the likelihood of replication fork collapse to form persistent specific DSB and inhibiting HR and NHEJ repair pathways (225–228). Therefore, based on the ability of PARPi to amplify unrepaired DNA damage, the combination of PARPi and RT has become an effective treatment method (67, 229–231).
Different preclinical studies have shown that the combination of RT and PARPi is beneficial in the treatment of BRCA wild-type cells. Preclinical studies have demonstrated that the PARPi 3-aminobenzamide (3-AB) can enhance radiosensitivity in BRCA wild-type cell lines by blocking the repair of radiation-induced SSB (232). Additionally, the combination of Olaparib and PI-103 has enhanced radiation-induced cell death in BRCA wild-type cells (165). Another preclinical study showed that the combination of Olaparib, RT, and ATRi (AZD6738) significantly inhibited the growth of HR-proficient tumors (233). However, to date, only a limited number of preclinical studies have provided insights into the therapeutic potential of combining PARPi and RT for the treatment of BRCA wild-type cancer. Further investigation into the mechanism of action of this combination therapy is still needed in clinical trials. In conclusion, all these preclinical trials suggest that PARPi combined with radiotherapy is a promising strategy to enhance tumor DNA damage. By increasing DSB load through, PARPi combined with RT can make BRCA wild-type cancer cells radiosensitive and promote the death of these cells.
7 ConclusionsAlthough PARPi have achieved great success in treating patients with BRCA1/2 mutated cancers, the current application of PARPi still faces some challenges, especially in improving the efficacy of PARPi in BRCA1/2 wild-type cancer patients and overcoming acquired resistance to PARPi. Currently, early trials combining PARPi with targeted drugs such as ATRi, WEE1i, and VEGFRi have shown some progress. Additionally, research on the combination of PARPi with DNA-damaging agents such as cytotoxic chemotherapy and radiation therapy are progressing, but there are challenges of cumulative toxicity when PARPi is used in combination with DNA-damaging agents. Finally, some trials found that PARPi combined with immunotherapy can enhance anti-tumor immune responses and improve treatment outcomes. In summary, this review outlines the basic principles and ongoing clinical trials (Table 1) of PARPi in combination therapy with various agents, expecting that the indications for PARPi will be optimized and expanded in the coming years.
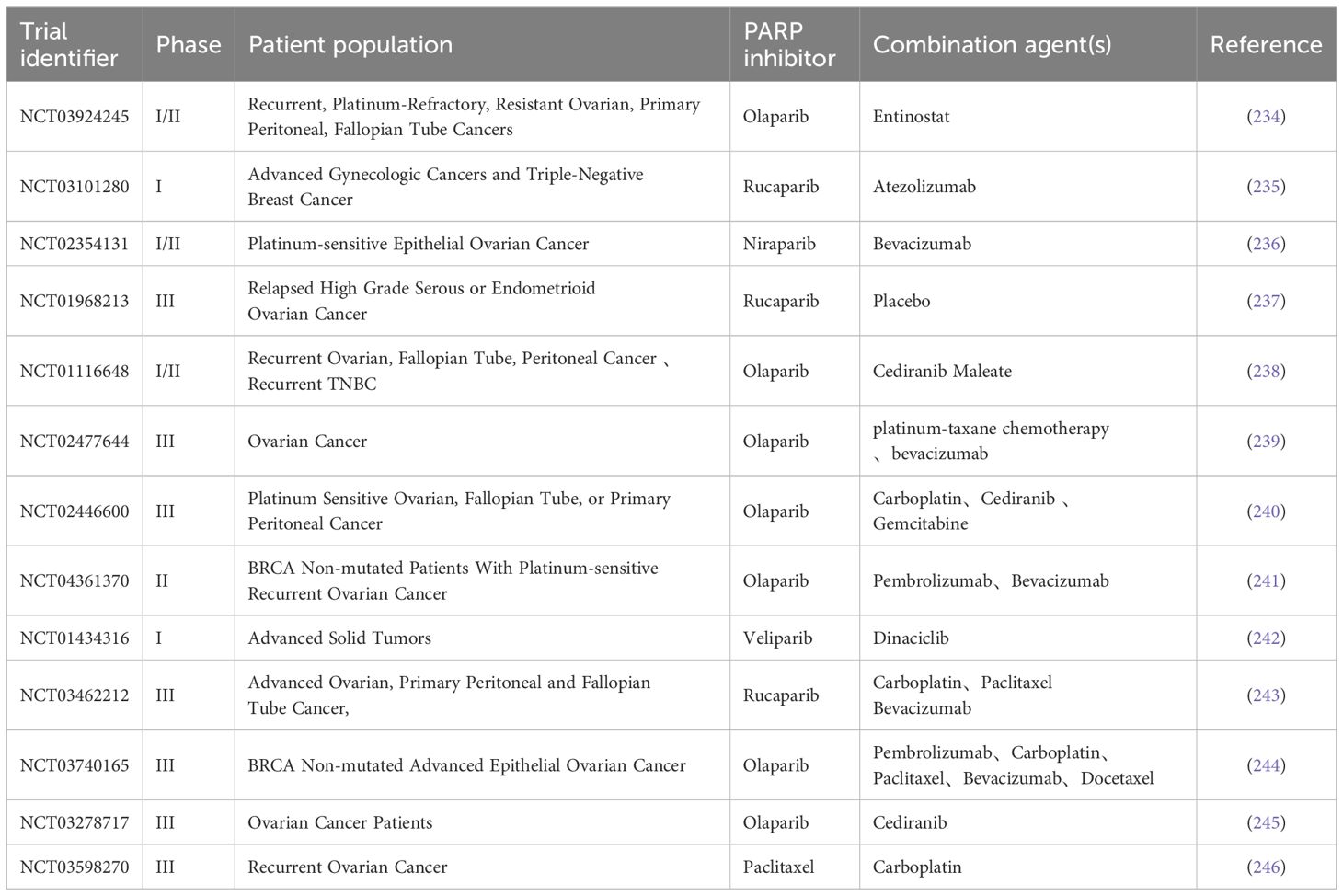
Table 1 Clinical trials in combination with PARPi for BRCA wild-type patients.
Author contributionsYX: Writing – original draft, Writing – review & editing. DX: Writing – original draft, Writing – review & editing. DL: Visualization, Writing – review & editing. MP: Visualization, Writing – review & editing. WP: Visualization, Writing – review & editing. HD: Writing – review & editing. XY: Writing – review & editing.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82172653); Institutional Open Fund (KF2022001);Key Project of Developmental Biology and Breeding from Hunan Province (2022XKQ0205), and The Research Team for Reproduction Health and Translational Medicine of Hunan Normal University (2023JC101).
AcknowledgmentsThis is a short text to acknowledge the contributions of specific colleagues, institutions, or agencies that aided the efforts of the authors.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. Sukhanova M, Khodyreva S, Lavrik O. Poly(Adp-ribose) polymerase 1 regulates activity of DNA polymerase beta in long patch base excision repair. Mutat Res. (2010) 685:80–9. doi: 10.1016/j.mrfmmm.2009.08.009
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Curtin NJ, Szabo C. Poly(Adp-ribose) polymerase inhibition: Past, present and future. Nat Rev Drug Discovery. (2020) 19:711–36. doi: 10.1038/s41573-020-0076-6
CrossRef Full Text | Google Scholar
3. Paul S, Sinha S, Kundu CN. Targeting cancer stem cells in the tumor microenvironment: An emerging role of parp inhibitors. Pharmacol Res. (2022) 184:106425. doi: 10.1016/j.phrs.2022.106425
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in brca mutant cells as a therapeutic strategy. Nature. (2005) 434:917–21. doi: 10.1038/nature03445
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of brca2-deficient tumours with inhibitors of poly(Adp-ribose) polymerase. Nature. (2005) 434:913–7. doi: 10.1038/nature03443
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Pilié PG, Gay CM, Byers LA, O'Connor MJ, Yap TA. Parp inhibitors: Extending benefit beyond brca-mutant cancers. Clin Cancer research: an Off J Am Assoc Cancer Res
留言 (0)