
記住我


The TRP superfamily of ion channels is composed of 28 members that are subdivided into six subfamilies in mammals. TRP channels are characterized by their low sequence homology and family clustering based on overall structural similarities (Clapham, 2003; Clapham et al., 2003; Venkatachalam and Montell, 2007; Cabezas-Bratesco et al., 2022).
The different TRP subfamilies are TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPA (ankyrin), TRPP (polycystin), and TRPML (mucolipin) (Ramsey et al., 2006). These families can be divided into two large groups, where TRPC, V, M, and A form the Class I group while polycystins and mucolipins form Class II group of TRP channels (Venkatachalam and Montell, 2007). Here, we will focus on the Class I type expressed in humans.
The first highlights in the discovery of TRP channels originated from observations of a phenotypic characteristic in a mutant strain of the fruit fly Drosophila (Cosens and Manning, 1969), where this mutant strain exhibited abnormal responses to light stimuli, indicating potential involvement of specific genes in sensory transduction processes. It was not until 1989 that Montell and Rubin assigned an identity using cDNA libraries obtained from mRNA extracted from the heads of adult Drosophila. They managed to identify the mRNA, which they named TRP (Montell and Rubin, 1989). Later, in 1992, they provided the first evidence that TRP proteins function as ion channels (Hardie and Minke, 1992).
TRP channels are tetrameric, non-selective, cation permeating channels structurally similar to voltage-gated potassium channels (Kalia and Swartz, 2013). Each TRP channel protomer comprises six transmembrane segments (S1–6), a pore region between S5 and S6, and intracellularly located N- and C-terminal domains, reviewed in (Cao, 2020; Zhang et al., 2023).
These channels can form homotetramers and/or heterotetramers depending on whether the subunits are identical or present one or more subunits from other TRP channels (Nilius, 2007; Gaudet, 2008; Cabezas-Bratesco et al., 2022). Furthermore, these channels interact with several accessory proteins that can regulate their expression and sub-cellular localization (Moiseenkova-Bell and Wensel, 2011; Cabezas-Bratesco et al., 2022; Cai and Chen, 2023).
Different mechanisms can regulate TRP channels, but in this review, we will focus on regulating the amount of TRP ion channels in the cell surface or trafficking to/from the plasma membrane (Royle and Murrell-Lagnado, 2003). In this context, the localization of an ion channel at the cell surface is determined partly by the balance between its synthesis and degradation, which occurs on a relatively slow time scale, as well as by its constitutive and regulated trafficking mechanisms (Bezzerides et al., 2004; Ambudkar, 2007; Dong et al., 2010; Toro et al., 2011). One of the main regulatory mechanisms of ion channels involves modulating the number of channel molecules expressed at the cell surface, either through the synthesis of new channels or the degradation of existing ones. However, if rapid regulation is needed, it can be achieved by either inserting or removing already synthesized channels stored in intracellular vesicles, which makes trafficking an important mechanism for regulating the activity of ion channels in a rapid fashion (Royle and Murrell-Lagnado, 2003). It is known that the expression of surface proteins can be regulated in various ways, one of which might be due to an increase in protein retention through its interaction with scaffold proteins during or after their insertion into the plasma membrane (Altschuler et al., 2003; Yu et al., 2011; Zhao et al., 2015; Van den Eynde et al., 2021). While the surface expression of ion channels requires the fusion of transporting vesicles mediated by the exocytic pathway, their internalization mechanisms require the activation of endocytic pathways (Royle and Murrell-Lagnado, 2003; Estadella et al., 2020).
Concerning the endocytosis of ion channels, the mechanism relies on various proteins and lipids that are present in specific domains of the plasma membrane, such as clathrin and caveolin, the soluble N-ethylmaleimide-sensitive factor attachment receptor protein (SNARE), or phosphoinositides such as PIP2, (Jahn et al., 2003; Laude and Prior, 2004). Different studies have shown that TRP channels traffic to specific cell regions to perform their function in polarized cells such as in the kidney, gastrointestinal tissue, or neurons. However, the mechanisms involved in this specific destination are still unclear for the most part (Altschuler et al., 2003; Yu et al., 2011; Zhao et al., 2015; Van den Eynde et al., 2021). Thus, a better understanding of the differential trafficking of these proteins could explain, for example, how TRPC6 reaches the basolateral and apical membranes of polarized epithelial cells. Also, TRPC3 is present on the apical side and TRPC1 is found mainly on the basolateral membrane (Singh et al., 2001; Bandyopadhyay et al., 2005; Goel et al., 2007).
This review will focus on the mechanisms reported for the trafficking and surface localization of the different class I TRP channels in mammalian cells, providing insights into the proteins involved in their subcellular targeting and trafficking. Moreover, as defects in the localization of ion channels have often been linked to the development of several pathologies, we will also provide an overview of diseases that have been related to the changes in the localization of these channels.
The TRPC subfamily of ion channels, insights into their trafficking and physiological significanceThe TRPC channel subfamily comprises seven members in mammals (TRPC1-7). TRPC2 is a human pseudogene and, therefore, was not considered in this review. Like other members of the TRP channel family (TRPA and TRPV), TRPC channels feature ankyrin repeat sequences within their N-terminal domain. Additionally, like the TRPM subfamily, these channels possess a proline-rich TRP structural domain located in the C-terminal region, close to the channel’s sixth transmembrane segment. On the other hand, in the N-terminal portion are three or four ankyrin repeats depending on the channel and a coiled-coil domain (Philipp et al., 2000; Lepage et al., 2006).
Like other ion channels, TRPC channels can form heterotetramers. For example, the TRPC1 channel can co-assemble with TRPC3-TRPC7 (Wu et al., 2004). TRPC channels can be activated through different mechanisms, such as Gq/11 receptors or receptor tyrosine kinase (RTK) downstream of the phospholipase C pathway (Trebak et al., 2007). Once phospholipase C (PLC) is activated, TRPC3/6/7 channels are activated by diacylglycerol (DAG) independently of protein kinase C (PKC), indicating that DAG mediates their physiological activation (Hofmann et al., 1999; Ma et al., 2000; Venkatachalam et al., 2002). On the other hand, TRPC1/4 and 5 channels are activated by receptor-induced PLC activation and are not responsive to DAG (Hofmann et al., 1999; Venkatachalam et al., 2003).
The function of TRPC channels is determined by their interactions with several proteins that affect their regulation, trafficking, and scaffolding, in addition to their effects on other downstream cellular processes (Ong and Ambudkar, 2011). It has been proposed that these interactions determine their localization and function in specialized plasma membrane microdomains (Ong and Ambudkar, 2011). TRPC channels have various conserved protein-protein interaction motifs in the N- and C-terminal regions, together with those that interact with WW-repeat domains, Cav1-scaffolding domains, PDZ-domains, and lipids such as PIP2. Moreover, they have ankyrin repeats and numerous coiled-coiled regions that appear particularly important in the assembly of the channels and their plasma membrane localization. Numerous proteins such as Homer, STIM1, Caveolin, NHERF, and PI3K, interact with specific TRPC channels and regulate their surface expression and channel activity (Table 1). Any alterations in these domains and interactions can result in dysregulation of the surface expression of the channels, contributing to the development of pathologies, reviewed in (Englisch et al., 2022).
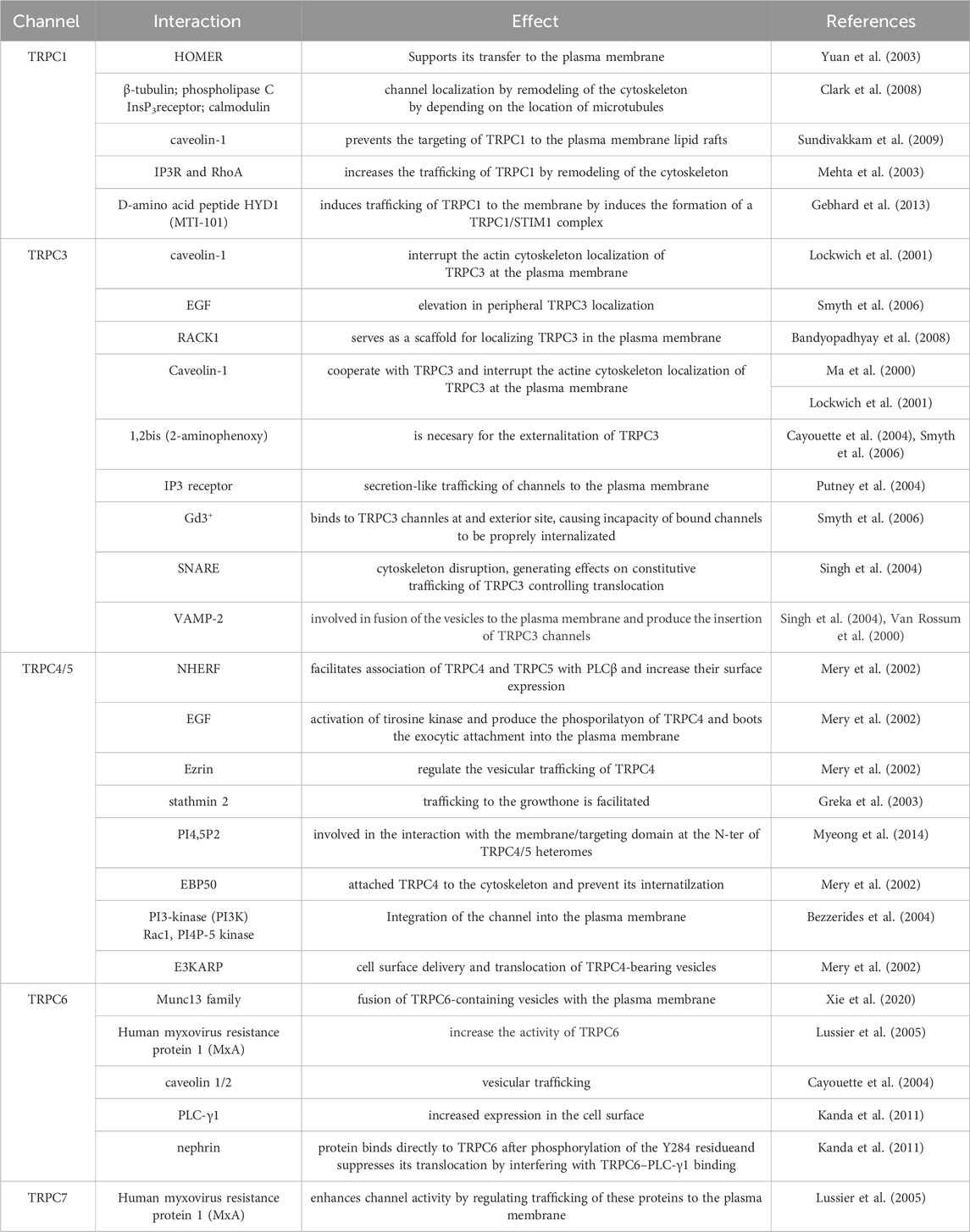
Table 1. Summary of TRPC trafficking interactions.
TRPC1TRPC1 is expressed in a large range of cell types and tissues. TRPC1 was originally found in the fetal brain, liver, and kidney tissues, as well as the adult heart, testes, ovaries, and brain (Wes et al., 1995). After these first descriptions, different groups have found the channel to be broadly expressed in mammalian tissues (Vazquez et al., 2004a; Beech, 2005; Ramsey et al., 2006; Ambudkar, 2013; Dietrich et al., 2014). Up to this point, TRPC1 performs a role in intracellular Na+ and Ca2+ homeostasis and although its function is not entirely clear (Hoth and Penner, 1992; Feske et al., 2006; Vig et al., 2006; Dietrich et al., 2014).
The trafficking of TRPC1 channels is controlled by several mechanisms involving interactions with lipid modifications, scaffolding proteins, and post-translational modifications (Ferrandiz-Huertas et al., 2014a). For instance, the interaction of TRPC1 with the scaffolding protein Homer has been shown to support its transfer to the plasma membrane, whereas palmitoylation of TRPC1 channels is involved in regulating their permanence and localization in the plasma membrane (Kiselyov et al., 1998; Torihashi et al., 2002; Yuan et al., 2003; Ambudkar et al., 2004). Moreover, TRPC1 localization is regulated by remodeling of the cytoskeleton through interaction with several proteins, including β-tubulin, IP3 receptor, calmodulin (CaM), PLC, and several other proteins (Clark et al., 2008). TRPC1 interacts with caveolin-1 through the C- and N-terminal domains. This interaction seems relevant in assembling a signaling complex localized in lipid rafts, important for regulating Ca2+ signaling (Sundivakkam et al., 2009).
The expression of an N terminal-truncated TRPC1 or a dominant-negative caveolin mutant prevents the TRPC1 trafficking to the plasma membrane affecting Ca2+ influx associated with direct activation of TRPC1 by agonists or passive store depletion. Additionally, in endothelial cells, trafficking of TRPC1 to the membrane occurs following thrombin stimulation (Mehta et al., 2003). Following activation, TRPC1 assembles in a complex with the IP3R and Ras homolog family member A (RhoA), a protein essential for the remodeling of the cytoskeleton (Liu et al., 2000; Lockwich et al., 2000; Brazer et al., 2003; Isshiki and Anderson, 2003; Mehta et al., 2003; Ambudkar et al., 2004; Brownlow et al., 2004; Bollimuntha et al., 2005). Impairments in TRPC1 channel trafficking have been related to various diseases, including cardiac hypertrophy. In ventricular myocytes, TRPC1 colocalized with sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 2 (SERCA2), an established marker of sarcoplasmic reticulum (SR), and sarcomeric α-actinin, which is a marker of the Z-line of the cardiomyocytes (Hu et al., 2020).
In this context, mutations in the TRPC1 gene have been identified in patients with familial essential hypertension (Jiang et al., 2014). Moreover, the abdominal aortic-banded model (AAB) in animals generates an upregulation of TRPC1 expression and an increased store-operated calcium entry (SOCE) in hypertrophied cardiomyocytes. In contrast, the knockdown of TRPC1 decreased SOCE. In this study, cardiomyocytes were treated with d-amino acid peptide HYD1 (MTI-101), which induces trafficking of TRPC1 to the membrane. Co-immunoprecipitation studies indicate that MTI-101 treatment induces the formation of a TRPC1- stromal interaction molecule 1 (STIM1) complex (Gebhard et al., 2013). This influx of Ca2+ activates the SOCE pathway and allows TRPC1 trafficking and insertion into the plasma membrane. Similar observations were made in NCI-H929 and U266 myeloma cell lines where aberrant trafficking of TRPC1 channels has been linked to the growth and metastasis of tumor cells (Gebhard et al., 2013; Myeong et al., 2018; Elzamzamy et al., 2020; Elzamzamy et al., 2021). Overall, the proper trafficking of TRPC1 channels to the membrane is crucial for its function in SOCE, and defects in trafficking can have profound effects on cellular physiology and human health.
TRPC3TRPC3 is a non-selective cation permeable channel that performs a critical role in several physiological activities, including cardiac hypertrophy, vascular smooth muscle contraction, and insulin secretion among others (Vazquez et al., 2004a). Various factors, including post-translational modifications and the activity of molecular chaperones through protein-protein interactions control this process (Li et al., 1999; Gonzalez-Cobos and Trebak, 2010). The proper localization of TRPC3 channels to the plasma membrane is vital for their function (Xia et al., 2015; Oda et al., 2017; Fan et al., 2018).
TRPC3 can be found assembled in a multimeric protein complex with key Ca2+ signaling proteins such as PLCβ, G alpha-q (Gαq) subunit of heterotrimeric G protein (Gαq/11), Inositol trisphosphate receptor (IP3Rs), CaM and PLCγ (Kiselyov et al., 1998; Lockwich et al., 2001; Zhang et al., 2001; Patterson et al., 2002; Wedel et al., 2003). However, the precise mechanisms governing TRPC3 trafficking to the plasma membrane remain unresolved.
Singh and colleagues proposed that agonist-induced activation of TRPCs also triggers exocytotic trafficking of the channel. They observed that carbachol (CCh) induces a vesicle-associated membrane protein 2 (VAMP-2)-dependent increase in the level of TRPC3 in the plasma membrane, independently of intracellular calcium concentration. TRPC3-containing vesicles were found to be positioned immediately beneath the plasma membrane, likely pre-docked to specific sites through the action of docking and scaffolding proteins. Moreover, they suggested that the surface expression of TRPC3 is regulated by both a recycling-type trafficking mechanism and a regulated event stimulated by CCh (Singh et al., 2004). Furthermore, in 2008, Bandyopadhyay and colleagues reported that the receptor for activated C-kinase-1 (RACK1) serves as a scaffold for localizing TRPC3 in the plasma membrane of cells. This interaction plays a crucial role in recruiting the channel into an IP3R-associated signaling complex and facilitating its insertion into the cell surface membrane upon stimulation by agonists (Bandyopadhyay et al., 2008).
In this context, CCh-induced exocytic trafficking of TRPC3 has been probed by surface biotinylation labeling showing that channel externalization occurs in the presence of the calcium chelator 1,2-bis (2- aminophenoxy) ethane-N,N,N,N-tetraacetic acid (BAPTA), reducing the possibility of a secondary effect by the Ca2+ mobilizing activity of the agonists (Cayouette et al., 2004; Smyth et al., 2006). On the other hand, several studies propose that TRPC3 trafficking is not mandatory for channel activation. They hypothesize that channel activation by second messengers, including DAG or conformational coupling of TRPC channels with the IP3R, could lead to secretion-like trafficking of channels to the plasma membrane (Vazquez et al., 2004b; Putney et al., 2004). Moreover, Smyth and colleagues demonstrated that Gd3+ binds to TRPC3 channels at an extracellular site, causing impaired internalization. Gd3+ effectively “locks” TRPC3 in the plasma membrane, favoring constitutive exocytosis over endocytosis and resulting in a swift accumulation of TRPC3 in the membrane. Furthermore, they observed an elevation in peripheral TRPC3 localization induced by epidermal growth factor (EGF), as assessed by total internal reflection fluorescence (TIRF) microscopy. This suggests that EGF may similarly stabilize TRPC3 in the plasma membrane by impeding its endocytosis, although the precise mechanism remains unclear (Smyth et al., 2006). Nonetheless, it is increasingly evident that the constitutive cycling of plasma membrane proteins is a fundamental mechanism for regulating surface membrane proteins (Royle and Murrell-Lagnado, 2003). TRPC3 also interacts with caveolin-1, a marker for caveolae, and disruption of the actin cytoskeleton deregulates the localization of caveolin-1 and TRPC3 at the plasma membrane (Ma et al., 2000; Lockwich et al., 2001).
Additionally, it has been reported that the N-terminal of TRPC3, specifically in amino acids 123-221, interacts directly with VAMP-2. VAMP-2 is located in the membrane of intracellular trafficking vesicles and mediates the fusion of vesicles to the plasma membrane, promoting the insertion of TRPC3 in the plasma membrane (van Rossum et al., 2000; Singh et al., 2004). Up to this point, the involvement of SNARE proteins in regulating TRPC3 localization at the plasma membrane is still not completely clear.
Importantly, atypical trafficking of TRPC3 channels can lead to diseases, including cardiac hypertrophy, hypertension, and Alzheimer’s disease (Tano et al., 2010; Smedlund et al., 2012; Tiapko and Groschner, 2018). It has been described that in cardiac hypertrophy, excessive trafficking of TRPC3 channels can lead to enhanced Ca2+ influx and stimulation of downstream signaling pathways like nuclear factor of activated T-cells (NFAT) and mammalian target of rapamycin (mTOR), leading to the development of hypertrophy (Brenner and Dolmetsch, 2007). These channels have been associated with regulating blood pressure, and changes in TRPC3 trafficking have been linked to pulmonary hypertension (Tano et al., 2010). In the brain, it is involved in the pathogenesis of Alzheimer’s disease, where Aβ peptides, which are linked to the development of Alzheimer’s disease, correlate with TRPC3-dependent calcium influx, leading to neurotoxicity (Montecinos-Oliva et al., 2014; Wang L. et al., 2017).
TRPC4/5TRPC4/5 channels are ubiquitously expressed in various tissues. They play a role in calcium signaling and ion homeostasis in various relevant physiological processes, including taste and vision, microvascular permeability, vasorelaxation, gastrointestinal motility, and neurotransmitter release (Fujita et al., 2017; Kim et al., 2019). Like many other membrane proteins, TRPC4 is synthesized in the ER, undergoing post-translational modifications, including glycosylation and phosphorylation. Then it is shipped to the Golgi apparatus, where it is further modified and sorted for delivery to the plasma membrane (Duan et al., 2018). Moreover, TRPC channels are particularly sensitive to mutations and deletions. These modifications may lead to reduced trafficking to the plasma membrane and their retention in the ER or Golgi (Xu et al., 2001; van de Graaf et al., 2003; Wedel et al., 2003; Andrade et al., 2005).
Additionally, two different studies showed that TRPC4 and TRPC5 interact with PDZ-domain-containing proteins such as the Na+/H+ exchanger regulatory factor (NHERF) and the gap junction protein zona occludens 1 (ZO-1) via their C-terminal PDZ-interacting domains (Weinman et al., 1995; Tang et al., 2000). The interface with NHERF facilitates the association of TRPC4 and TRPC5 with PLCβ, which, in turn, positively regulates their surface expression (Mery et al., 2002). Moreover, cellular stimulation by EGF activates protein tyrosine kinases, leading to the specific phosphorylation of two tyrosine residues located at the C-terminal end of TRPC4 (Mery et al., 2002). Such post-translational modification boosts the interaction of TRPC4 and NHERF and the plasma membrane expression of the channel (Mery et al., 2002).
TRPC4 is regulated by the growth factor receptor signaling proteins, such as ezrin, in addition to NHERF binding proteins, which regulate the vesicular trafficking of TRPC4 (Mery et al., 2002). In this context, it has been observed that TRPC5 is trafficked to certain sites in hippocampal neurons and that TRPC5 homomers are found in growth cones (Greka et al., 2003). Trafficking of TRPC5 to the growth cone is facilitated via attaching to the exocytic protein stathmin 2 (a protein associated with ALS), where Ca2+ influx through the channel prevents the extension of growth cones. On the other hand, SNARE proteins interact with the TRPC5 trafficking complex (Greka et al., 2003). TRPC5 channels are localized in vesicles, and fast trafficking of these vesicles to the plasma membrane is triggered in response to stimulation with EGF and nerve growth factor (NGF) and a faint response to brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-1 (IGF-1). Integration of the channel into the plasma membrane also involves Phosphoinositide 3-kinase (PI3K) and the GTPase Rac Family Small GTPase 1 (Rac1), as well as PI4P-5 kinase (Bezzerides et al., 2004). Additionally, TRPC5-including vesicles seem to be retained in a subplasma membrane region from where they are rapidly recruited to the membrane (Ambudkar, 2013).
Mery et al. (2002) showed that the proximal N-terminal region of TRPC4 (amino acids between 23 and 29 in the mice ortholog) is vital for membrane insertion of TRPC4 channels. Altering this domain might cause a decrease in trafficking to the plasma membrane by preventing either the appropriate folding or heterotetrameric assembly with wild-type TRPC4 (Mery et al., 2002). Myeong et al. (2014) demonstrated that the N- and C-terminal regions interact with each other to make a tetrameric structure and that PI(4,5)P2 is a strong candidate involved in the interaction with the membrane-targeting domain at the N terminus of TRPC4/5 heteromers. They reported that the N terminal (amino acids 98–124) and C-terminal (amino acids 700–728) were necessary for the tetrameric assembly of TRPC4 (Myeong et al., 2014; Hong et al., 2015). The interaction between the scaffold ERM-binding phosphoprotein 50 (EBP50) (the NHERF human ortholog) and the membrane-cytoskeleton adaptors can attach TRPC4 to the cytoskeleton and prevent its internalization. NHE3 kinase A regulatory protein (E3KARP), a member of the ezrin/radixin/moesin (ERM) family, plays an active role in the cell surface delivery of TRPC4 by enabling the translocation of TRPC4-bearing vesicles from the cortical actin layer to the plasma membrane. Removal of the last three C-terminal amino acids in TRPC4 blocks the interaction with EBP50 and alters the channel’s localization and surface expression (Mery et al., 2002). In the same study, the authors suggested that TRPC4 can reach the submembranous compartment but is not delivered to the cell surface (Mery et al., 2002).
Dysregulation of TRPC4/5 trafficking and/or function can lead to various diseases (Myeong et al., 2014) in the brain (Mori et al., 1998; Fowler et al., 2007), peripheral sensory neurons (Wu et al., 2008), and gastrointestinal organs. Moreover, a few articles have implicated TRPC4 in ocular diseases, including retinitis pigmentosa and glaucoma (Yang et al., 2005; Reinach et al., 2015; Yang et al., 2022). In retinitis pigmentosa, mutations in TRPC4 result in abnormal protein trafficking to the plasma membrane, leading to photoreceptor cell death and vision loss. In glaucoma, the dysregulation of TRPC4 trafficking is thought to contribute to the death of retinal ganglion cells, which leads to optic nerve damage and vision loss (Yang et al., 2005; Reinach et al., 2015; Yang et al., 2022).
TRPC4 is also associated with several cardiovascular diseases, including cardiac hypertrophy, heart failure, and hypertension. TRPC4 plays a crucial role in regulating the heart’s electrical activity, and abnormal trafficking of TRPC4 can lead to dysregulation of these processes. Mutations in the TRPC4 gene have been associated with atrial fibrillation, Brugada syndrome, and long QT syndrome, whereas TRPC5 has been linked to the development of cardiac hypertrophy and heart failure, as well as pulmonary hypertension (Watanabe et al., 2008; Camacho et al., 2015; Lüscher, 2015; Du et al., 2021). TRPC5 regulates Ca2+ signaling and vascular smooth muscle tone, crucial cardiovascular physiology processes. Impaired TRPC4 trafficking can lead to calcium overload in cardiomyocytes, which dysregulates their function and can lead to heart dysfunction. TRPC4 is also involved in regulating vascular smooth muscle tone, and alterations in its activity can lead to increased blood pressure and hypertension (Watanabe et al., 2008; Camacho et al., 2015; Lüscher, 2015; Du et al., 2021).
TRPC4 has been implicated in several neurological disorders, including multiple sclerosis and Parkinson’s disease, due to its involvement in the regulation of calcium signaling and neuronal excitability (Balbuena et al., 2012; Wang L.-K. et al., 2017; Enders et al., 2020). On the other hand, TRPC5 has been linked to depression, anxiety, and drug addiction (Yang et al., 2005; Nazıroglu and Demirdas, 2015; Yang et al., 2015; Sharma and Hopkins, 2019). Studies have shown that TRPC5 regulates the release of neurotransmitters and modulates synaptic plasticity, which are important processes in the pathogenesis of these disorders (Puram et al., 2011; He et al., 2012). Recent studies have shown that TRPC4 regulates the volume of airway epithelial cells, and its dysfunction may contribute to the pathogenesis of cystic fibrosis (Müller et al., 2022). TRPC5 is predominantly expressed in the renal system and has been linked to the pathogenesis of several renal diseases, including focal segmental glomerulosclerosis (FSGS), diabetic nephropathy, and polycystic kidney disease (Zhou et al., 2017; Yu et al., 2019; Walsh et al., 2021).
TRPC6Several factors can affect the TRPC6 expression on the plasma membrane of channels in non-excitable cells. For instance, the stimulation of endogenous muscarinic receptors with CCh and the depletion of intracellular calcium stores generate the translocation of TRPC6 toward the plasma membrane (Cayouette et al., 2004). There is still no clear evidence of the participation of SNARE proteins in the trafficking of TRPC6. It has been reported that the Human myxovirus resistance protein 1 (MxA), closely related to the membrane-remodeling GTPase dynamin, interacts with TRPC6 (Lussier et al., 2005). Among the proteins involved in TRPC6 trafficking induced by DAG stimulation, the mammalian uncoordinated-13 (Munc13) family of proteins participates in the fusion of TRPC6-containing vesicles with the plasma membrane, providing insights into the DAG-regulated vesicle fusion mechanism (Xie et al., 2020).
Regarding the glycosylation of TRPC6, it has been described that it is glycosylated in residues N474 and N561. The removal of glycosylation in residue N561 is sufficient to convert it into a constitutively active channel, while trafficking and surface expression remain unaffected in glycosylation-deficient mutants (Dietrich et al., 2003; Talbot et al., 2019).
As mentioned above, TRPC6 can form heterotetramers with other members of the TRPC channel family. Three independent studies observed that once activated, TRPC6 is translocated to the plasma membrane along with TRPC3 (Cayouette et al., 2004; Singh et al., 2004; Kim et al., 2006). In another study, HEK293 cells were stimulated with CCh, resulting in the translocation of TRPC6 in the caveolae, which are closely linked to vesicular traffic (Cayouette et al., 2004; Cayouette and Boulay, 2007). Interestingly, this translocation mechanism is independent of intracellular calcium increases (Stojilkovic et al., 2005).
Moreover, another mechanism involved in TRPC6 trafficking was reported by Kanda et al. (2011), who demonstrated that phosphorylation of a single residue (Y284 in the mice ortholog) is required for channel surface expression. Moreover, a tyrosine to phenylalanine mutation (Y284F) showed decreased expression on the cell surface of both HEK293T cells podocyte cultures, suggesting that the residue is necessary for PLC-γ1 binding (Kanda et al., 2011). In addition, the authors observed that in podocytes, the nephrin protein binds directly to TRPC6 after phosphorylation of Y284, suggesting that a naturally occurring mutation in this position might be associated with focal segmental glomerulosclerosis disease (FSGS) (Kanda et al., 2011). Coincidently, Hagmann and colleagues (2018) have proposed that phosphorylation at S14 in mice TRPC6 enhances channel conductance by increasing the membrane expression of TRPC6 (Hagmann et al., 2018).
TRPC7The TRPC7 channel is the most recently identified member of the TRPC family, and little is known about the mechanisms governing its trafficking to the plasma membrane. Additionally, there is not much information about naturally occurring mutations that can affect the channel’s expression and/or trafficking. Lussier et al. (2005) demonstrated through different approaches, including GST-pull-down and co-immunoprecipitation assays, that MxA interacts with the second ankyrin-like repeat domain of TRPC7 and other TRP channels. It has been proposed that MxA enhances channel activity by regulating trafficking to the plasma membrane (Lussier et al., 2005). However, this functional role seen in other TRP channels has not been tested on TRPC7 yet.
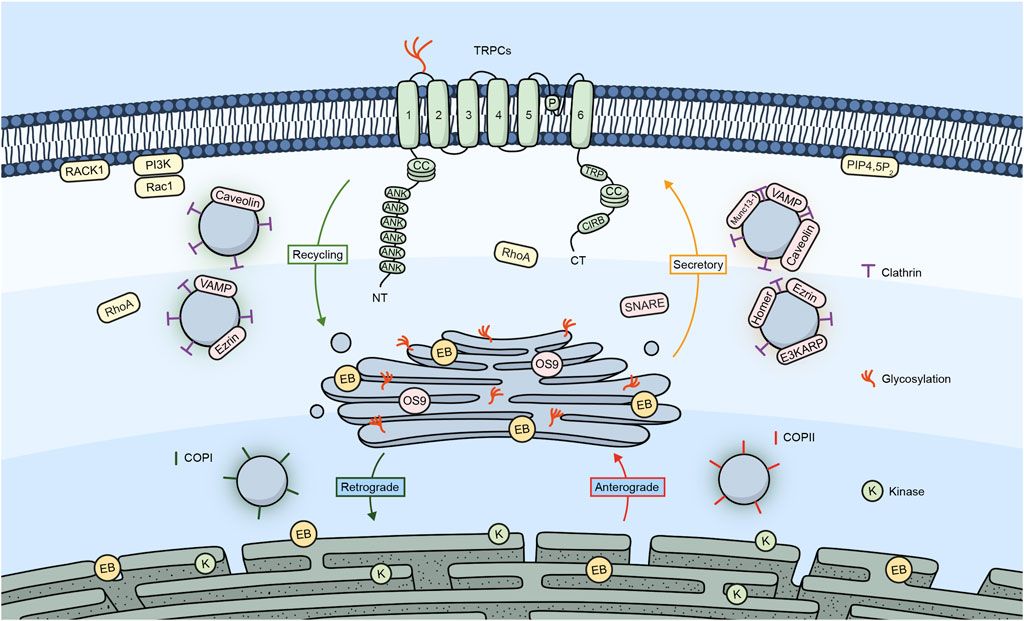
Figure 1 shows a summary of the different proteins/molecules involved in the regulation of the trafficking of TRPC channels to the plasma membrane as were described in this section of the review.
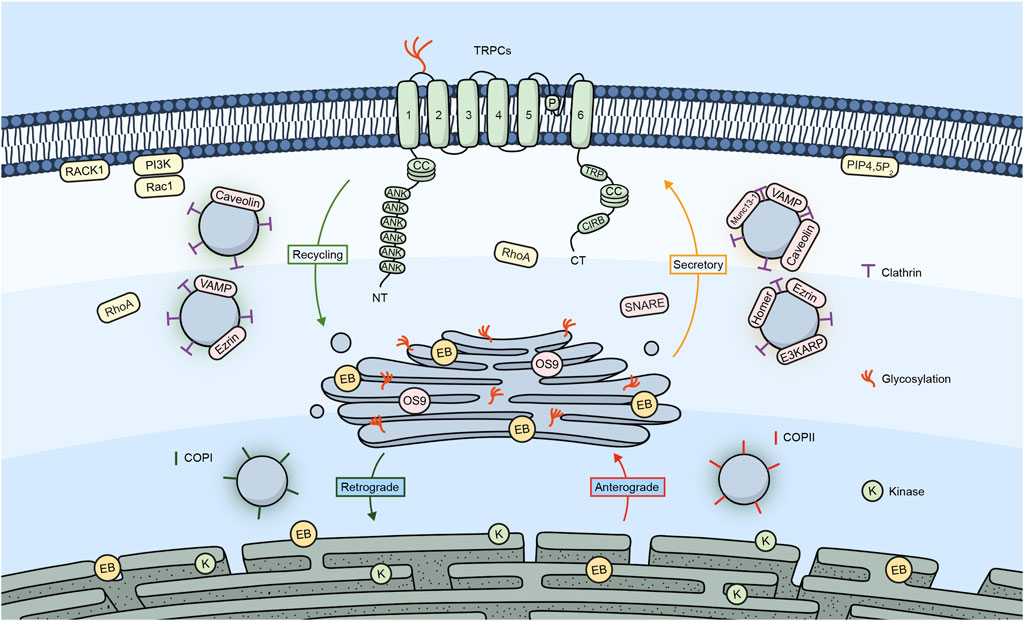
Figure 1. TRPC channels and their trafficking regulators. Embedded in the plasma membrane is a monomeric subunit representing the various domains and regions in TRPC channels. These include ANK (ankyrin repeats), CC (coiled-coil), TRP, which refers to the TRP box common to all TRP channels and a calmodulin, and IP3R binding site (CIRB). The diagram shows a summary of the different regulators involved in the trafficking of TRPC channels.
The TRPM subfamily of ion channels, insights into their trafficking and physiological significanceThe human TRPM subfamily is the largest among those belonging to the superfamily of TRP channels and was named after the founding member Melastatin (TRPM1) (Fleig and Penner, 2004). Since the first description and cloning of TRPM1 in 1998 (Duncan et al., 1998), seven more members have been added to this subfamily, which can be grouped into four pairs based on their structural similarity (TRPM1/TRPM3; TRPM2/TRPM8; TRPM4/TRPM5; and TRPM6/TRPM7) (Cohen and Moiseenkova-Bell, 2014). As the rest of the TRP channels, TRPMs are intrinsic membrane proteins with six transmembrane domains that form functional tetramers. TRPMs are widely expressed in the human body. Most TRPMs are Ca2+-permeable cation channels, except for TRPM4 and TRPM5, which are Ca2+-activated, non-selective, monovalent permeable cation channels (Chen et al., 2019; Yamaguchi et al., 2019). The members of the TRPM subfamily of ion channels are characterized by their polymodal nature of activation as they are regulated by different stimuli such as voltage, temperature, and the binding of ions, lipids, or other ligands (Ramon et al., 2007; Diaz-Franulic et al., 2021).
The TRPM subfamily has garnered greater attention due to its reported involvement in several physiological processes, such as taste transduction (Liu and Liman, 2003), temperature sensing (McKemy et al., 2002; Brauchi et al., 2004), synaptogenesis and neurite outgrowth (Abumaria et al., 2019), cell death (McNulty and Fonfria, 2005), and regulation of vasculature (McNulty and Fonfria, 2005). Moreover, the abnormal expression and/or function of TRPM channels has also been involved in a plethora of pathological processes such as cancer (Hantute-Ghesquier et al., 2018; Saldías et al., 2021), neurological disorders (Sita et al., 2018; Lavanderos et al., 2020), kidney disorders (Hsu et al., 2007), and others (Jimenez et al., 2020). All this poses TRPM channels and their regulation as important therapeutic targets.
In the following section, we will discuss the main mechanisms involved in the trafficking of TRPM channels and how the dysregulation of this process might trigger different pathologies. Like TRPC channels, the trafficking of TRPM channels is regulated by specific protein interactions summarized in Table 2.
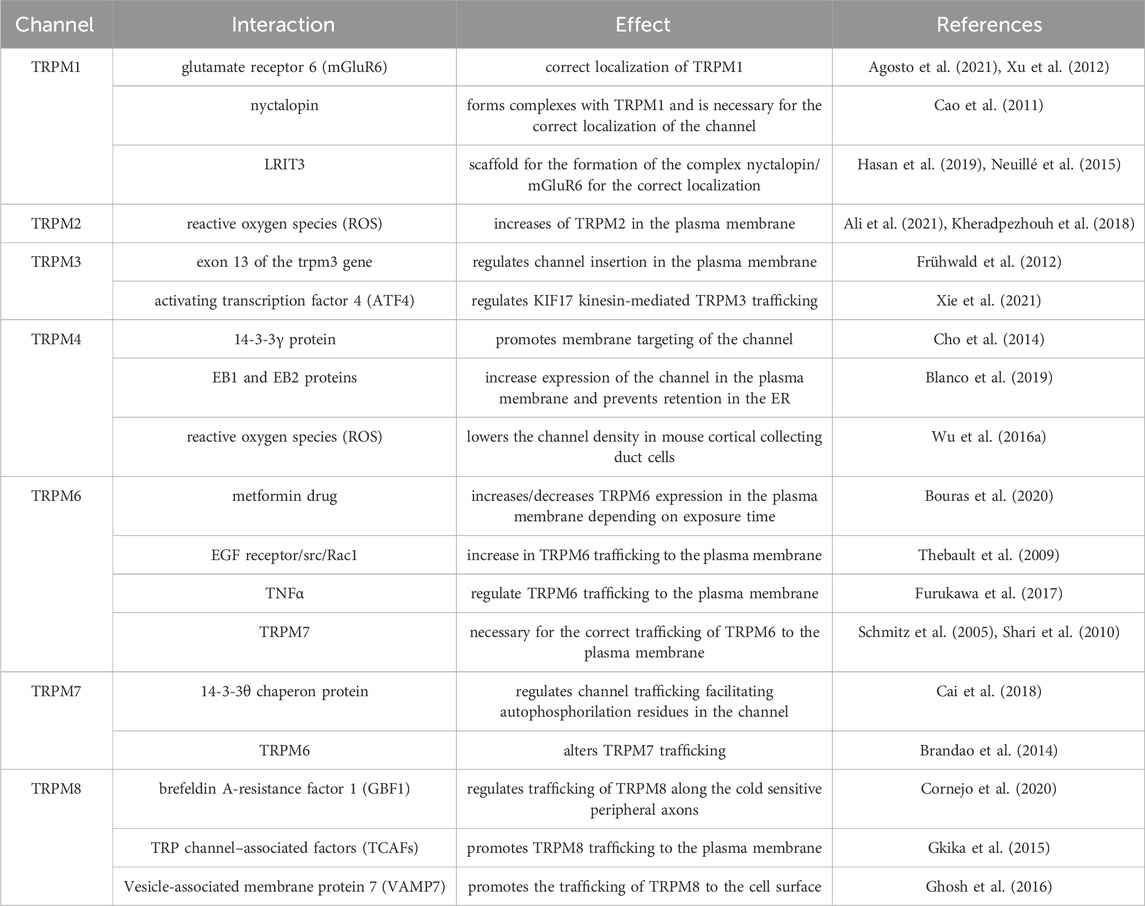
Table 2. Summary of TRPM trafficking interactions.
TRPM1TRPM1 received the name Melastatin due to its initial discovery in the B16 mouse melanoma cell line (Duncan et al., 1998). TRPM1 expression is not as ubiquitous as with other TRPM channels. Its expression has been reported in the heart, brain, skin, and retina (Fonfria et al., 2006; Oancea and Wicks, 2011). As mentioned before, TRPM1 is involved in Melanoma progression, where expression of TRPM1 inversely correlates with tumor aggressiveness and tumor thickness (Deeds et al., 2000; Guo et al., 2012). It has also been linked to disease-free survival in patients with melanoma (Brozyna et al., 2017), suggesting that TRPM1 may function as a metastatic suppressor. Additionally, it has been reported that TRPM1 is the ion channel that initiates the ON visual pathway in vertebrate vision in the eye (Koike et al., 2010; Nakamura et al., 2010; Iosifidis et al., 2022). TRPM1 trafficking mechanisms have not been fully described yet, but a few reports have suggested possible mechanisms. Nakamura and colleagues showed in ON bipolar cells that mutations R624C and F1075S lead to lower expression in the dendritic tips and mislocalization of TRPM1, which translates to stationary night blindness in patients. These results suggest that these residues are important for the protein trafficking of TRPM1 (Nakamura et al., 2010). Interestingly, the metabotropic glutamate receptor 6 (mGluR6), specifically its C-terminal region, appears to have an important role in the correct localization of TRPM1 to the dendritic tips of depolarizing bipolar cells, as the mGluR6 knockout and other mutants show reduced to no expression of TRPM1 in this region (Xu et al., 2012; Agosto et al., 2021). Moreover, it has been demonstrated that nyctalopin also forms complexes with TRPM1 and is necessary for the correct channel localization (Cao et al., 2011). Additionally, another molecule that has been involved in the trafficking of TRPM1 and the formation of its complex with nyctalopin and mGluR6 is Leucine Rich Repeat, Ig-Like And Transmembrane Domains 3 (LRIT3), which might serve as a scaffold for the formation of this complex since the knockout mouse model for this protein showed no localization of TRPM1 at the dendritic spines of ON bipolar cells (Neuillé et al., 2015; Hasan et al., 2019). These reports suggest that mutations in the interacting residues of TRPM1 with proteins might result in channel trafficking defects.
TRPM2TRPM2 is a ligand-gated Ca2+-permeable non-selective cationic channel. Despite having a structure that is very similar to the rest of the TRPM subfamily, TRPM2 possesses a C-terminal Nudix hydrolase 9 homologue (NUDT9-H) domain that serves as a binding site for cytosolic adenosine diphosphate ribose (ADPR) (Huang et al., 2018). The gating mechanism of TRPM2 channels seems to require simultaneous co-activation by Calcium (Csanády and Törocsik, 2009), PIP2 (Tóth and Csanády, 2012), and ADPR (Perraud et al., 2001; Hara et al., 2002). Although TRPM2 contains this NUDT9-H domain, it has little to no ADPR-degrading ability and would only act as a regulatory ligand binding site (Iordanov et al., 2016). In terms of expression, TRPM2 is nearly ubiquitous in all human tissues and, thus, has been implicated in regulating several physiological and pathological processes (Belrose and Jackson, 2018). TRPM2 plays an important role in migration and chemokine release (Yamamoto et al., 2008). TRPM2 modulates cell migration and invasion in neuroblastoma, and its expression has been associated with poor patient prognosis (Bao et al., 2022), whereas it regulates autophagy during replication of the Hepatitis B virus (Chen et al., 2022). Moreover, it has also been involved in regulating cell death in several cell models (Malko et al., 2019; Zielinska et al., 2021) and as a potential therapeutic target for treating neurological diseases (Sita et al., 2018; Malko et al., 2019).
About trafficking, the most studied regulatory mechanism of TRPM2 is the effect that reactive oxygen species (ROS) have over its localization. Reports have shown that ROS increase TRPM2 activity (Ali et al., 2021) and treatments with either H2O2 or the non-opioid analgesic acetaminophen (both leading to the production of ROS) increase TRPM2 in the plasma membrane (Kheradpezhouh et al., 2018). Moreover, Mei and Jiang reported that although they saw no effect for the N-terminal coiled-coil domain of TRPM2 in the trafficking of the channel, they could not discard a minor contribution of this domain to the transport of the channel (Mei and Jiang, 2009). Additionally, the expression of a short splicing variant of TRPM2 decreased the channel’s activity, mostly by decreasing the trafficking of the full-length channel to the plasma membrane (Yamamoto et al., 2019).
TRPM3TRPM3 is a Ca2+ permeable, non-selective cation channel activated by heat and chemical activators which, interestingly, shares certain homology with the heat sensitive TRPV channels (Vriens et al., 2011). As with other TRPM channels, TRPM3 is expressed in a wide variety of tissues in the body (Held et al., 2015), with a relatively higher expression in the brain, spinal cord, sensory neurons, pituitary, kidney and eye (Fonfria et al., 2006; Shaham et al., 2013; Oberwinkler and Philipp, 2014). TRPM3 plays an important role in pancreatic β-cells, where it regulates zinc uptake (Wagner et al., 2010), and in the eye, where its mutations lead to cataracts and glaucoma (Bennett et al., 2014; Zhou et al., 2021). However, its most reported role is the one it plays as a nociceptor in detecting noxious heat and developing inflammatory heat hypersensitivity (Held et al., 2015; Mickle et al., 2015; Held and Tóth, 2021).
The mechanisms involved in TRPM3 trafficking are not well understood. However, we have some information from the few works that have addressed the topic. It has been reported that an 18 amino acid sequence encoded by exon 13 of the trpm3 gene regulates channel insertion in the plasma membrane and is key for sustaining TRPM3 activity (Frühwald et al., 2012). Moreover, it has been recently proposed that mutations in the N-terminal part of the protein, specifically the L769V mutation, affect trafficking to the plasma membrane and result in a non-functional channel (Burglen et al., 2023). Additionally, it has been reported that the activating transcription factor 4 (ATF4), a member of the CREB family of proteins, regulates kinesin family member 17 (KIF17) -mediated TRPM3 trafficking by interacting with KIF17-TRPM3 complex and modulating dorsal root ganglia (DRG) neuron response to heat stimuli (Xie et al., 2021).
TRPM4TRPM4 differs from most other TRP channels by not being permeable to Ca2+ but only to monovalent cations (showing a high selectivity for Na+). However, one of its main features is that intracellular Ca2+ can directly activate it (Launay et al., 2002; Ullrich et al., 2005). TRPM4 is expressed widely throughout the different body tissues, although several reports have shown a higher expression level in the colon, heart, and prostate (Launay et al., 2002; Fonfria et al., 2006; Syam et al., 2016; Borgstrom et al., 2021). Moreover, TRPM4 expression has been reported in several other cell types, such as immune cells, neuronal tissue, and pancreatic β-cells (Cheng et al., 2007; Barbet et al., 2008; Schattling et al., 2012). This wide expression pattern underlines the important role this channel might play in several physiological and pathological processes. TRPM4 has been reported to play a role in pathologies such as different types of cancer (Rivas et al., 2020; Borgstrom et al., 2021; Wang et al., 2022), and cardiovascular (Stallmeyer et al., 2012; Syam et al., 2016; Wang et al., 2018) and immune system-associated diseases (Barbet et al., 2008; Schattling et al., 2012; Serafini et al., 2012).
Trafficking mechanisms of TRPM4, unlike other TRPM channels, have been more widely studied in recent years. Cho et al. (2014) demonstrated that membrane targeting of the channel is regulated by interaction with the 14-3-3γ protein through the N-terminal region of TRPM4 and that S88 in the human ortholog, which might be a target of phosphorylation, is key for 14-3-3γ binding and anterograde trafficking of TRPM4 (Cho et al., 2014). On the other hand, in patients with human progressive familial heart block type I, the E7K mutation at the N-terminal domain caused attenuated deSUMOylation of the channel, which impaired channel endocytosis and led to an increased activity of TRPM4 due to an elevated concentration of the channel in the plasma membrane (Kruse et al., 2009). Interestingly, several other mutations showed a similar effect in patients with isolated cardiac conduction. Mutations R164W, A432T, and G844D enhance TRPM4 current density due to increased channel abundance in the membrane caused by SUMOylation defects and impaired endocytosis (Liu et al., 2010). Moreover, in patients with congenital or childhood atrioventricular block, mutations A432T and A432T/G582S showed decreased protein expression at the cell membrane, whereas G582S alone showed increased membrane expression of the channel (Syam et al., 2016). Although N-glycosylation also plays an important role in the surface expression and trafficking of ion channels, its role in TRPM4 trafficking is somehow controversial. Several reports showed that the N988 residue in rats and N992/N932 residues in humans are glycosylation sites for TRPM4, but these mutations did not affect the surface expression of the channel but rather modulated its stability (Woo et al., 2013; Syam et al., 2014). Interestingly, phosphorylation also appears to have a role in TRPM4 trafficking. Phosphorylation of S839 is necessary for basolateral localization of TRPM4 in epithelial cells (Cerda et al., 2015). Other mutations have been reported to impact TRPM4 trafficking without an apparent change in the electrophysiological properties of the channel, such as those described in Brugada syndrome, where P779R and K914X showed a decreased membrane expression whereas T873I and L1075P showed an increase in membrane localization of TRPM4 (Liu et al., 2013). Another mechanism reported for TRPM4 anterograde trafficking is its interaction with end-binding (EB) proteins. Blanco et al. (2019) showed that mutations in the EB binding motif in TRPM4 prevented its interaction with EB proteins, which led to a reduced expression of the channel in the plasma membrane and retention in the ER (Blanco et al., 2019). Interestingly, TRPM4 localization in the plasma membrane is also modulated by ROS, given that hydrogen peroxide treatment lowers the channel density in mouse cortical collecting duct cells (Wu M. et al., 2016).
TRPM5TRPM5, like TRPM4, is a nonselective monovalent cation channel impermeable to divalent cations activated by Ca2+ and has a high permeability for Na+ (Prawitt et al., 2003). However, unlike TRPM4, which is widely expressed in several tissues and organs, TRPM5 expression is more limited to highly specialized cells. For example, high expression of the channel has been reported in β cells from pancreatic islets of Langerhans, where it regulates the frequency of Ca2+ oscillations that lead to glucose-stimulated insulin secretion by β-cells (Brixel et al., 2010; Ketterer et al., 2011). Moreover, its most iconic role is played in taste-sensing receptor cells, where it is co-expressed with other receptors and signaling molecules involved in the process of bitter, sweet, and umami stimuli detection (Perez et al., 2002; Zhang et al., 2003; Dutta et al., 2018). TRPM5 expression has also been reported in other chemosensory cells in the olfactory, respiratory, and digestive systems, suggesting a broader role in chemosensory processes (Kaske et al., 2007; Bezencon et al., 2008; Lin et al., 2008). The available information regarding TRPM5 trafficking is insufficient to relate this process to specific cellular or tissue failure. However, unlike what has been reported for surface proteins (Khanna et al., 2001), the N-glycosylation would not be involved in the trafficking of TRPM5 to the plasma membrane but rather regulates channel function (Syam et al., 2014).
TRPM6TRPM6 is a Ca2+ permeable ion channel, which is also highly permeable to Mg2+ (Schlingmann et al., 2007). Interestingly, it exhibits an unusual feature, as it contains a transmembrane segment fused to a cytosolic α-type serine/threonine protein kinase domain, which allows it to act as a chanzyme (Chubanov et al., 2018). TRPM6 expression is more restricted than most TRPM channels, with the highest levels of expression reported in the kidney and the gastrointestinal tract, where it is, interestingly, regulated by dietary magnesium (Groenestege et al., 2006; Lameris et al., 2015). The most important role of TRPM6 is related to the homeostasis of systemic Mg2+ homeostasis, where different mutations and dysregulation of its activity have been widely reported in patients with different forms of hypomagnesemia (Schlingmann et al., 2002; Chubanov et al., 2007; Lainez et al., 2014) while also being important for the correct functioning of colonic epithelial cells (Luongo et al., 2018).
It has been reported that patients with hypomagnesemia with secondary hypocalcemia present the S141L mutation, which causes a decrease in the expression of TRPM6 at the plasma membrane (Chubanov et al., 2004). Moreover, Bouras et al. (2020) showed that short-term exposure to type 2 diabetes mellitus drug metformin (1,1-dimethylbiguanide) increased TRPM6 expression in the plasma membrane while, interestingly, long-term exposure significantly decreased the plasma membrane expression of the channel (Bouras et al., 2020). Another interesting mechanism reported for TRPM6 trafficking suggests activating the EGF receptor increased TRPM6 trafficking to the plasma membrane through the src kinase-Rac1 pathway (Thebault et al., 2009). Moreover, Furukawa et al. suggested that Tumor necrosis factor α (TNFα) could regulate TRPM6 trafficking to the plasma membrane (Furukawa et al., 2017). Additionally, although phosphorylation by the close relative TRPM7 and TRPM6/TRPM7 multimers does not appear to alter TRPM6 trafficking (Brandao et al., 2014), it appears TRPM7 is still necessary for the correct trafficking of TRPM6 to the plasma membrane (Schmitz et al., 2005; Shari et al., 2010).
TRPM7TRPM7, although being discovered independently, is very similar in structure and function to its homolog TRPM6, being also a Mg2+/Ca2+ permeable ion channel (Nadler et al., 2001; Runnels et al., 2001; Schmitz et al., 2003). As with TRPM6, it contains the unique C-terminal serine/threonine protein kinase domain, which shares a low homology with other kinases and is important for the autoregulation of channel activity (Schlingmann et al., 2007; Cabezas-Bratesco et al., 2015; Chubanov et al., 2018). Unlike TRPM6, TRPM7 is widely expressed in many tissues and organs (Nadler et al., 2001; Runnels et al., 2001) but a higher expression has been reported in the heart and kidney (Montell, 2005).
Given its ubiquitous expression, TRPM7 has been involved in several physiological and pathological processes (Visser et al., 2014). For example, in the brain, it regulates synaptic transmission and plasticity, while on the other hand, it also regulates neuronal death following ischemia or neuron injury (Aarts et al., 2003; Krapivinsky et al., 2006; Abumaria et al., 2018). In this context, the fusion of synaptic-like vesicles in PC12 cells is somewhat modulated by vesicular TRPM7 activity (Brauchi et al., 2008). Moreover, it has been reported in several types of cancer, such as breast (Guilbert et al., 2013) and ovarian cancer (Wang et al., 2014), as a metastasis promoter. In contrast, in the heart, it has been involved in forming cardiac fibrosis (Yu et al., 2014).
In the context of TRPM7 trafficking, its most studied regulator is its close homolog, TRPM6. It has been reported that the TRPM6 kinase domain can phosphorylate TRPM7 in serine residues, resulting in altered TRPM7 trafficking. However, TRPM7 phosphorylation of TRPM6 showed no effect on its localization (Schmitz et al., 2005; Brandao et al., 2014).
Interestingly, another regulator of TRPM7 trafficking is TRPM7 itself. It has been shown that S1360 of TRPM7 is a key residue for autophosphorylation, mediating both TRPM7 stability and intracellular trafficking. Other important autophosphorylation residues in the channel were S1403 and S1567, whose autophosphorylation by TRPM7’s kinase activity regulates its interaction with the 14-3-3θ chaperon protein, which regulates channel trafficking (Cai et al., 2018).
TRPM7 channels experience two types of trafficking. The complete gene product traffics to the plasma membrane, where a proteolytic cleavage product containing the kinase domain (M7CKs) is generated and translocated to the nucleus. These fragments establish interactions with proteins composing chromatin remodeling complexes (Krapivinsky et al., 2014). This mechanism is relevant during development (Desai et al., 2012), and the transient overexpression of mouse TRPM7 in HEK293 cells was found to significantly alter the cellular transcription of hundreds of genes (Lee et al., 2011). Finally, it has been shown that sheer stress on blood vessels can increase TRPM7 in the plasma membrane (Oancea et al., 2006).
TRPM8TRPM8 is a non-selective Ca2+-permeable channel activated by cold, membrane depolarization, and different cooling compounds and is considered the most important thermoreceptor in cold perception (McKemy et al., 2002; Brauchi et al., 2004; Bautista et al., 2007). TRPM8 was initially identified in prostate tissue and reported as upregulated in prostate cancer (Tsavaler et al., 2001). The cold current was first identified in DRG sensory neurons (Reid and Flonta, 2001), and then, the channel was identified, cloned, and described (McKemy et al., 2002).
Although mainly expressed in sensory neurons, channel expression has also been reported in several other tissues and organs, such as the cardiovascular system, lungs, bladder, and urogenital tract (Dhaka et al., 2008; Iftinca and Altier, 2020; Liu et al., 2020). Growing evidence has linked TRPM8 expression and function to several types of cancer, including prostate, lung, and breast cancer (Fuessel et al., 2003; Dhennin-Duthille et al., 2011; Hantute-Ghesquier et al., 2018). Additionally, it has also been involved in the development of neuropathic pain (Proudfoot et al., 2006; De Caro et al., 2019) and irritable bowel syndrome (de Jong et al., 2015).
As one of the most studied TRP channels, there’s a wide variety of articles regarding TRPM8 trafficking and membrane context. It was initially reported that TRPM8 channels are located in lipid rafts (Morenilla-Palao et al., 2009). These overexpressed TRPM8 channels reside in a near membrane compartment, actively recycling in and out of the plasma membrane where its motility is restricted (Veliz et al., 2010; Ghosh et al., 2014). This recycling is associated with the SNARE protein VAMP7 (Ghosh et al., 2016). These recycling modes have been shown to vary in response to channel activation, modulating the number of available channels at the plasma membra
留言 (0)