In recent decades, nanotechnology-based therapeutic products have been introduced in clinical studies, and several nanomedicines have successfully entered the market. Nowadays, new health technologies (NHTs) and innovative therapeutic agents are being developed, marking an era of scientific and technological evolution [1].
Virtual reality (VR), artificial intelligence (AI), robotic surgery, 3D and bioprinting, digital therapy, medical devices, wearables, and bioinformatics stand out as important tools in the development of innovative therapies, contributing significantly to the advancement of precision and personalized medicine. Regulatory agencies face challenges in accurately defining the structural characteristics of nanocarriers and in adapting and scientifically managing the concept of complexity in the analysis of their properties. Off-patent nanomedicines should adhere to precise and accurate criteria established by the regulatory agencies to describe nanocarriers and these criteria should cover the physicochemical, thermodynamic, morphological, and biological properties, of nanocarriers, including surface interactions and pharmacokinetics. The term "nanosimilar" is the most appropriate for describing off-patent nanomedicines seeking regulatory approval as ‘similars’ to prototypes.
The non-linear and chaotic behavior exhibited by self-assembled nanocarriers in nanomedicinal products and in off-patent nanomedicines that are promoted as nanosimilars, necessitates from the regulatory point of view, a groundbreaking conceptualization with precise terminology and a framework for studying and approving dossiers submitted by stakeholders. Such approaches should parallel scientific and technological developments and motivate regulatory authorities to build a more reliable approval process.
Complexity at Nanoscale
Nanotechnological platforms are self-assembled nanostructures primarily composed of individual amphiphilic molecules that trigger new properties in the final nanostructure. These are all characterized as complex nanosystems [2].
Biological complex entities, such as proteins, can be mixed with amphiphilic molecules to produce nanostructures which modify their pharmacokinetic properties and consequently their effectiveness. Such behavior enhances their nonlinearity and their distance from equilibrium, and consequently changes the entropic profile of the nanocarrier and drives complexity towards a more functional morphology [3].
Artificial cell membranes like liposomes or lipid nanoparticles follow the above concept. At the next level of complexity, artificial cell membranes self-assemble and create the most predominant and effective nanostructures in terms of morphological, biophysical, and thermodynamical properties. Such properties have been ‘selected’ to surpass a threshold and acquire the qualitative information needed to become the fittest intermediate nanostructure for the next step of evolution. Such complex nanostructures may yield new properties unique to nanocarrier, which affect the effectiveness of the final off-patent nanomedicines and its approval as nanosimilar. However, these properties are challenging to reproduce due to their complex and chaotic behavior, while their cryptic ‘decision-making’ algorithm should be disclosed using advanced techniques [4]. The lipidic domains on the surface, known as ‘lipid rafts’, coexist in both the internal and surface part of the nanostructure. Such ‘lipid rafts’ can be considered as information hubs [5] that affect both the short and long-range order of the self-assembled complex nanocarrier and, as described previously, should be extensively evaluated by the regulatory agencies through a reliable process in order to be considered as nanosimilars.
Reproducibility and scaling up the production of nanomedicines and nanosimilar products,, poses specific challenges:
Interfacial Phenomena Characterization: Understanding and determining the interfacial interactions between the nanocarrier’s surface and the surrounding environment (e.g. pH, ionic strength, osmolarity etc.) are crucial for ensuring reproducibility.
Functionality and Stimuli Responsiveness Assessment: The preciseevaluation of the functionality and responsiveness to stimuli (e.g., temperature, pH, etc.) in the final nanomedicine and in the off-patent nanomedicine, termed as nanosimilar product, is essential for the intended therapeutic applications.
Nanosystem Complexity Analysis: The complexity of the nanocarrier, including the role of surface lipid rafts, (i.e., ‘lipid rafts’) [5] as well as the internal nanolipidic domains, should be evaluated.
Dynamic Phenomena Identification: Non-linear, dynamic, chaotic, thermodynamic, and morphological phenomena during the self-assembly process are key factors influencing the evolution of the nanocarrier and the final nanomedicine and nanosimilar product and they should be evaluated.
Addressing all these aspects will contribute to a more reliable scientific environment and to controlled mass production of nanostructures, fostering accuracy in the development process, and facilitating comprehensive discussions between stakeholders and regulatory authorities. The European Pharmacopoeia has established a working party on mRNA vaccines, and papers published for public comment in Pharmeuropa are used to ensure the quality of nanomedicines and their follow-on products (https://www.edqm.eu/en/-l/ph.-eur.-commission-establishes-a-new-working-party-on-mrna-vaccines, https://www.edqm.eu/en/-/edqm-event-quality-requirements-for-nanomedicines).
The off-patent nanotechnological therapeutic products belong to the grey area, which is named as mesoscopic, and the nanocarriers are called mesoscopic systems [6] and may qualify as ASAEs, a term associated with their complexity, non-linear and chaotic behavior. These principles are not only valid for ASAEs, but also for complex nanoparticulate APIs such as iron complexes.
The terms ‘similar’ and ‘similarity’ need to be clarified when characterizing off-patent nanomedicines as nanosimilars. A new regulatory framework needs to be established to facilitate discussions with stakeholders to develop safe and effective off-patent nanomedicinal products. Generic medicinal products are approved through bioequivalence studies, while biosimilars, due to their complexity and immunogenicity, follow the concept of clinical studies. These are important concerns in the manufacturing process [7, 8]. The quotes and concepts regarding the scientific tools that are needed for establishing a sustainable, dynamic, and multifunctional approval process for nanomedicine and nanosimilars have been published by Demetzos and co-workers [9]. The development of assessment procedures concerning ‘similarity’ in terms of physicochemical thermodynamical, morphological, and biological properties, as well as surface coating and surface properties, and the interfacial phenomena in biological media, should be properly considered. The pharmacokinetics, biodistribution, stability, surface interactions with other biomolecules, and potential outcomes of these interactions are all impacted by the complexity of short and long -range order, and the nonlinear and chaotic behavior of nanocarriers, which must also be considered in the evaluation process by the regulatory authorities.
Nanomedicines are categorized as Non-Biological Complex Drugs (NBCDs; in our opinion this needs correction to NBCMedicines) and the quotes from the published work [9] could be advised to find out a reliable, scientific, sustainable, dynamic and multifactional approval process. Klein et al. [10] present the views of the NBCD working group and solutions for approving complex generics by optimizing FDA pathways 505(j) and 505(b)(2), aiming to enhance regulatory clarity. The FDA defines complex generic drugs as those with complex active ingredients, formulations, routes of delivery, or dosage forms. This classification aims to facilitate the development of NBCDs, and to enhance the accessibility and affordability [10]. FDA guidance as well as reflection papers published by the European Medicines Agency (EMA) help bring together academic scientists, stakeholders, and regulatory authorities to discuss issues and create a dynamic regulatory environment. In our view, the existing regulatory environment requires improvements to clarify the complexity of promoting off-patent nanomedicines as nanosimilars. Artificial Intelligence (AI) startup companies must collaborate with big Pharma stakeholders to develop deep neural networks (DNNs). The internal complexity of the nanocarrier, varies, even during the development process in batch-to-batch production. It is important to schedule a complex large-scale system in pharmaceutical sector to study the complexity and to incorporate complex nanoplatfroms in a broader theoretical framework leading to the precise and accurate production of complex nanocarriers as drug delivery platforms. Such approaches guide regulatory agencies in a most realistic scientific and regulatory environment that facilitates effective appointments with stakeholders to discuss in depth the submitted NDA (New Drug Application) of the nanomedicine or of the nanosimilar products to be approved [11]. Cloud platforms incorporate libraries and data of diverse nanomaterials with comprehensive morphological and biological entities, enabling correlation with potential adverse drug effects and with the toxicity of advanced excipients. However, AI could provide valuable support in expending the existing scientific and regulatory process for discussing and evaluating the properties of nanoparticles due to their complexity and to their non-linear behavior, overcoming barriers current regulatory approaches. Recently published work by Wong, and colleagues (Wong, F et al., 2024) [12], refers to the discovery of a structural class of antibiotics with explainable deep learning. To sum up, the lack of precise nomenclature, manufacturing issues, quality control concerns, scalability complexities and regulatory issues constitute, in our view, obstacles to the establishment of a regulatory framework embracing and facilitating the approval process for off-patent medicines as nanosimilars.
Off-Patent Nanomedicines in the Market. Regulatory Issues
The FDA has recognized that off-patent nanomedicines already in the market, serving as follow-on versions of the prototype nanomedicinal formulations and approved as generic versions of the prototype nanomedicine, adhere to the bioequivalence concept.
Lipodox® comprises doxorubicin hydrochloride encapsulated in long-circulating pegylated liposomes, FDA-approved in 2013 as a generic of the anticancer medicine Doxil® and /or Caelyx® which are the prototype nanomedicines of doxorubicin HCl in the USA and the European Union (EU), respectively. In the EU, such off-patent nanomedicinal products have been approved as ‘hybrid-medicines’ due to the absence of established essential criteria for promoting them as nanosimilars. Zolsketil® (Accord Healthcare S.L.U., Marketing-authorization holder) consists of pegylated liposomes encapsulating doxorubicin, the active substance of Doxil® and Caelyx®. It is categorized as a “hybrid medicine” and was approved by EMA in 2023 because it is viewed as comparable to Adriamycin and bioequivalent to Doxil® and Caelyx®.
Celdoxome® pegylated liposomal was approved in 2023, as a “hybrid medicine” encapsulating doxorubicin, the active substance, into a pegylated liposomal formulation. According to the EMA, and in accordance with EU requirements, it has been shown that the reference drug (i.e., doxorubicin/ adriamycin) is similar and the final product is bioequivalent to the pegylated liposomal formulation of the prototype nanomedicine, Caelyx®.
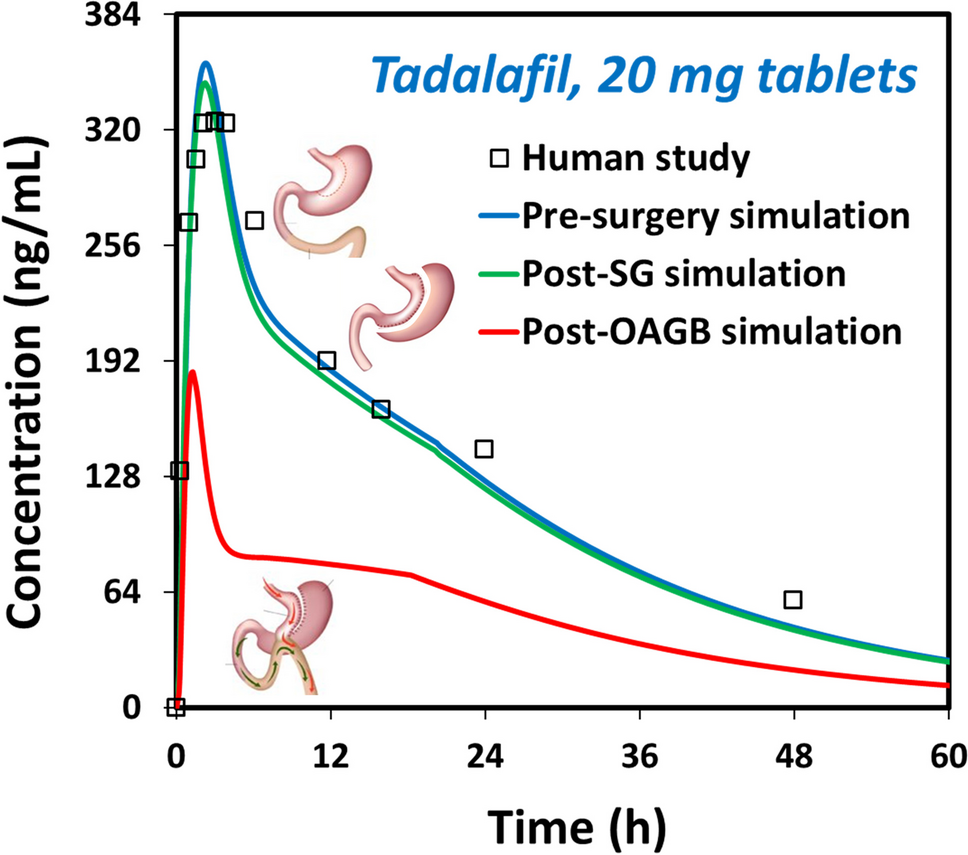
The field of nanomedicinal products should also be expanded to include the frequently used iron intravenous (IV) therapy. Iron nanomedicines consist of an iron core surrounded by a carbohydrate shell [13]. Such complexes are classified as non-biological complex drugs that cannot be fully characterized using only their physicochemical properties such as size, size distribution, polydispersity index, ζ-potential. Mehedinti and co-authors [13], aimed to address the issue that ‘there is no appropriate regulatory evaluation system for these medicines and several follow-on versions of the IV iron originators (e.g., iron sucrose) were approved using the same regulatory pathway as for generics’. The authors aimed to underline ‘the importance of intravenous iron therapy as well as raise awareness regarding the differences between nanomedicines and their intended similar but not identical copies’ and also emphasized that ‘The potential implications of these differences impact patients (safety, efficacy) but also the medical system (higher costs)’. It is of interest that Venofer®, which is the iron sucrose originator product cannot be completely characterized only by physicochemical analysis and needs essential criteria for characterization. Both clinical and non-clinical studies between iron sucrose similars (ISS) and the reference product showed no interchangeability [13].
Funk and co-authors [14] aimed to highlight that ‘The surface chemistry differences between the iron–carbohydrate complexes result in significant differences in in vivo pharmacokinetic and pharmacodynamic profiles as well as adverse event profiles, demonstrating that the entire iron–carbohydrate complex furnishes the pharmacologic action for these complex products. Currently available physicochemical characterization methods have limitations in biorelevant matrices resulting in challenges in defining critical quality attributes for surface characteristics for this class of complex nanomedicines’.
In a recently published paper, Klein and co-authors, explore the recent regulatory environment regarding the approval process of NBCDs [15]. The authors aimed to determine whether NBCDs approval follows a non-centralized authorization concept in EU leading to heterogeneity in the regulatory approach. NBCDs are not considered as a special category of medicines but there is no defined common regulatory pathway in the EU.
Bioinformatics, cheminformatics, and medical informatics have paved the way to facilitate the determination of nanotoxicity regarding the nanocarriers of therapeutic products. Such approaches facilitate regulatory agencies in the evaluation process of innovative therapeutic nanoformulations and in checking the role of nanoparticulate carriers, as their complexity demands. Robust physicochemical characterization data are important for the pharmaceutical industry to understand the unique properties of nanoparticles and of the final nanomedicine and to correlate them with biological performance. Regulatory authorities should supplied with such information to enhance their understanding of the unique physicochemical profile of nanomedicines in connection with the clinical performance. Such integrated approaches will facilitate filling the gap between scientific and regulatory parties and will help developers and regulatory authorities to ensure new communication pathways.







留言 (0)