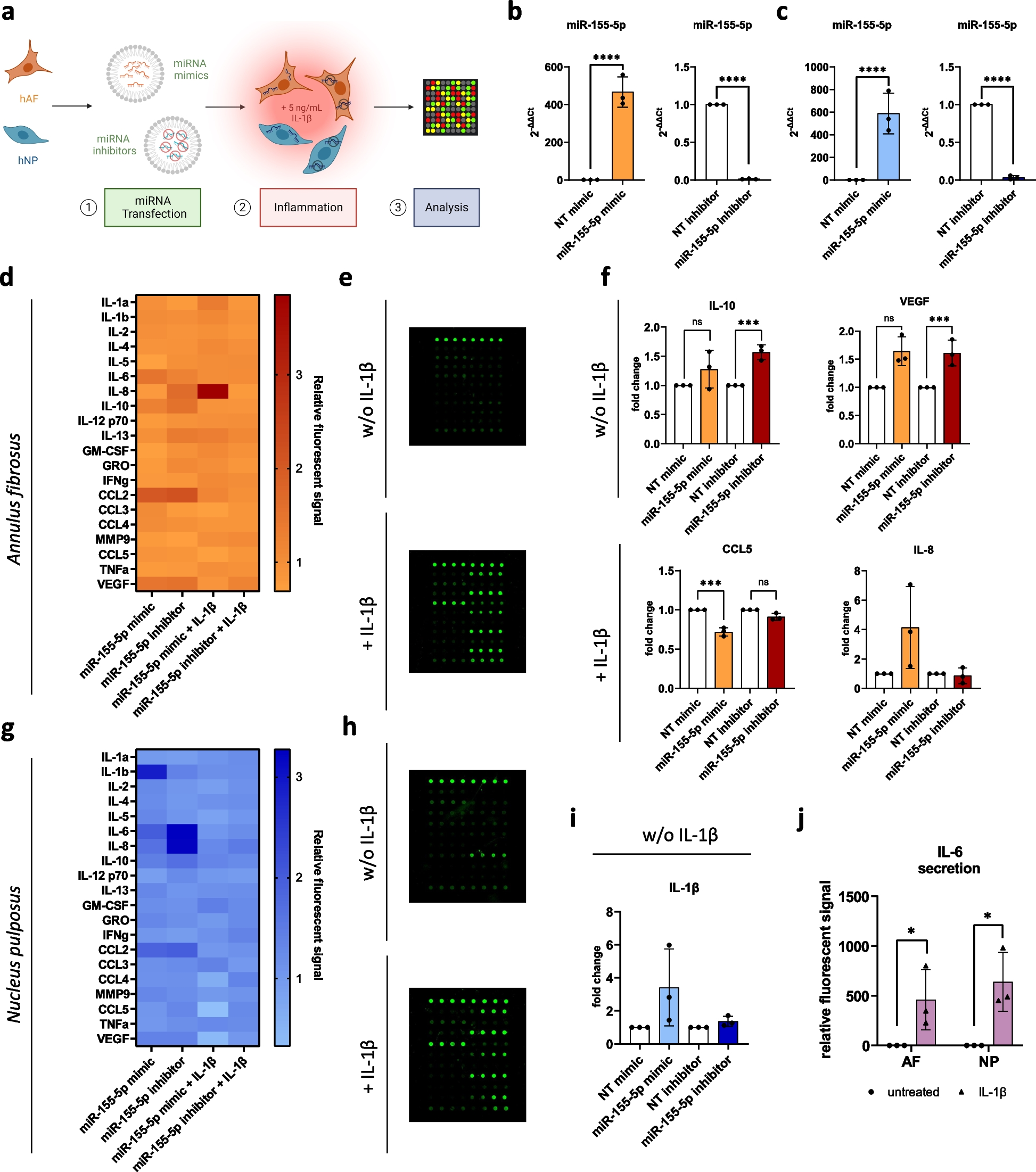
Cell culture
We cultured immortalized human endometrial glandular cells obtained from Kanazawa University’s Department of Obstetrics and Gynecology according to a previously described method [30]. We refer to this cell line as “hEM” below. The cells were cultured at 37 °C under 5% CO2 in DMEM/Ham’s-F12 (1:1) medium containing ITS (#I3146, Sigma–Aldrich, St. Louis, MO, USA), 10% fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 µg/ml) (#15140-122, Gibco/Thermo Fisher Scientific, Waltham, MA, USA).
Lentiviral production
We used Precision LentiORF IL17RB w/ Stop Codon (#OHS5897-202618086, Horizon Discovery, Waterbeach, UK) as the lentiviral expression vector, with a IL17RB insert. We used a Precision LentiORF RFP positive control (#OHS5832, Horizon Discovery) as the control vector. The lentiviral vectors were packaged and then transduced into HEK293T cells according to a previously described method [31] using Lipofectamine 2000 (#11,668,019, Thermo Fisher Scientific). Forty-eight hours after initiation of the transduction, we collected the culture supernatant and passed it through a 0.45 μm filter, and then added the lentivirus-containing filtrate to hEM cultures in the presence of polybrene, to a final concentration of 8 ng/µl.
Protein extraction and western blotting
We washed the cells in phosphate-buffered saline (PBS) cooled them to 4 °C, then lysed them in lysis buffer (#C2978, Sigma–Aldrich) containing a protease inhibitor cocktail (#P8340, Sigma–Aldrich) and phosphatase inhibitor cocktail (#07574-61, Nacalai Tesque, Kyoto, Japan). Electrophoresis was run in 10% precast gradient polyacrylamide gels (5 µg protein per lane). We transferred the separated proteins from the gels onto nitrocellulose membranes at 20 V overnight, after which we blocked the membranes either for 20 min with 5% milk or for 1 h with polyvinylidene fluoride (PVDF) blocking reagent (#NYPBR01, Toyobo, Osaka, Japan). We added the following primary antibodies and allowed the samples to react overnight at 4 °C: IL17RB (1:1000, #GTX127368, GeneTex, Irvine, CA, USA), Phospho-IκBα (Ser32) (14D4) mAb (1:1000, #2859, Cell Signaling Technology, Danvers, MA, USA), IκBα (44D4) mAb (1:1000, #4812, Cell Signaling Technology), Phospho-SAPK/JNK (Thr183/Try185) mAb (1:2000, #9255, Cell Signaling Technology), SAPK/JNK mAb (1:1000, #9252, Cell Signaling Technology), GAPDH (1:5000, #015-25473, Fujifilm Wako Pure Chemical Corporation, Osaka, Japan). Next, we washed the membranes with TBST (a mixture of tris-buffered saline and polysorbate 20) and then reacted them with one of the following secondary antibodies for 1 h at room temperature: Anti-Rabbit or Anti-Mouse IgG (whole molecule)-peroxidase antibody produced in goat (1:5000, #A6154 or #A4416, Sigma–Aldrich). We detected specific protein bands using SuperSignal West Dura Chemiluminescent Substrate (#34,080, Thermo Fisher Scientific).
NF-κB reporter assay
We simultaneously transfected the cells with pNL3.2 NF-κB-RE vector (#N1111, Promega, Madison, WI, USA) and pGL4.53 (Luc2/PGK) vector (#E5011, Promega) using FuGENE HD Transfection Reagent (#E2313, Promega). We seeded them in 96-well plates 24 h after transfection and, after another 24 h, treated them with IL17B at one of the following concentrations: 0, 20, or 200 ng/ml. We evaluated luciferase activity 6 h after IL17B treatment using the Nano-Glo Dual-Luciferase Reporter Assay System (#N1610, Promega) and a VICTOR Nivo Multimode Microplate Reader (PerkinElmer, Waltham, MA, USA).
RNA purification and RT-qPCR
We purified RNA samples using a RNeasy Plus Mini Kit (QIAGEN, North Rhine-Westphalia, Germany) and subjected them to a one- or two-step quantitative reverse transcription–polymerase chain reaction (RT-qPCR). For the two-step RT-qPCR, we performed the reverse transcription using ReverTra Ace qPCR RT Master Mix (#FSQ-201, Toyobo) and real-time PCR using specific primers for each gene (Table 1). We ran the experiments using TB Green premix Ex Taq TM II (#RR820A, Takara, Shiga, Japan) or a SYBR Green RT-PCR kit (#204,243, QIAGEN) on the CFX Connect Real-Time PCR System (BioRad, Hercules, CA, USA) operated according to the manufacturer’s protocol. We quantified the expression levels of each target gene relative to those of endogenous controls (HPRT-1, 18SrRNA) using the ∆∆CT method (control expression: 1.0).
Table 1 Primers used for RT–PCRThree-dimensional organoid cultures derived from human endometrial tissue
We prepared three-dimensional (3D) organoid cultures from human endometrial tissue donated by hysterectomy patients. This model is hereafter referred to as “patient-derived endometrial organoids (EOs)”. All donors gave their written informed consent, and their background information is shown in Table 2. These experiments were approved by the institutional review board of Kyushu University (approval no. 22087-00). To examine the in vivo morphology of the tissue, we fixed part of each specimen in formalin, embedded it in paraffin, sliced it into 4 μm sections, and stained these with hematoxylin–eosin (HE) for histological observation via optical microscopy. We collected the rest of the tissue in 4 °C PBS, centrifuged it at 2000 × g for 6 min to remove the supernatant, and then treated it with lysis buffer to remove erythrocytes. We prepared an enzymatic mixture by adding DNase (#2270A, Takara) and 10 µM Y-27,632 dihydrochloride (#034-24024, Fujifilm Wako Pure Chemical Corporation) to 5 mg/ml collagenase type II (#LS004197, Worthington Biochemical Corporation, Lakewood, NJ, USA) in HBSS (#17461-05, Nacalai Tesque). We treated the pellet from the previous step with this enzymatic mixture at 37 °C for 1–1.5 h, agitating it by pipette every 15 min. Next, we used 100 μm filters (#93,100, SPL Life Sciences, Gyeonggi-do, Korea) and TrypLE Express (#12604-021, Gibco) to disaggregate the tissue into individual cells and collect epithelial cells.
Table 2 Background of hysterectomy patientsWe pipetted the isolated cells onto a 24-well plate at a seed density of 3.5 × 104 cells per 10 µL Matrigel (#356,231, Corning, Corning, NY, USA). We then cultured the cells in organoid expansion medium (“ExM”) prepared according to a previously described method [38] with replacement every 2–3 d (500 µl/well; composition shown in Table 3).
Table 3 Compotision of basal medium and expansion mediumWe examined organoid morphology via HE staining. We stripped each culture of Matrigel by adding Cell Recovery Solution (#354,253, Corning), pipette-agitated it, washed it with basal medium, and then centrifuged them to prepare a pellet. The result was jellified using iPGell (Nippon Genetics Corporation, Tokyo, Japan), according to the manufacturer’s protocol, followed by fixation in 4% paraformaldehyde. The solidified suspension was then embedded in paraffin, sliced into 4 μm sections, HE-stained, and observed under an optical microscope.
Organoid-forming efficiency analysis
We treated the cultured organoids with Cell Recovery Solution to remove the Matrigel, agitated them by pipetting, and trypsinized them using TrypLE Express. We counted the disaggregated cells using a hematocytometer grid and then seeded them on a 24-well plate at a density of 3.5 × 104 cells per 10 µl Matrigel. To each well, we added 500 µl ExM containing one of three recombinant human cytokines: IL6 (#200-06, PeproTech, Cranbury, NJ, USA), IL8 (#200-08, PeproTech), or IL1β (#200-01B, PeproTech). We maintained the cultures for 10–20 d, exchanging the cytokine-containing medium every 48 h. Finally, we observed them using an all-in-one fluorescence microscope (BZ-X710: Keyence, Osaka, Japan) to check for cytokine-associated differences in organoid-forming efficiency, which we quantified as the number of organoids (i.e., ≥ 20 μm in diameter) per unit volume in Z-stacked images (2.28 × 10−1mm3) using BZ-X analysis software (BZ-X700 Analyzer, Keyence).
SA-β-gal assay
We measured senescence-associated β-galactosidase (“SA-β-gal”) using a SPiDER-β-Gal detection kit (#SG-03, Dojindo, Kumamoto, Japan) according to the manufacturer’s protocol. Organoids seeded and cultured in a 48-well plate were immuno-labeled after ExM removal and washing. We added Cell Recovery Solution to remove the Matrigel, and then dispersed the organoids via pipette agitation and trypsinized them using TrypLE Express. We washed the samples and passed them through a 40 μm filter; dead cells were removed using propidium iodide (#P-4170, Sigma–Aldrich). We measured SPiDER-β-Gal fluorescence using a BD FACSMelody Cell Sorter (BD Biosciences, Franklin Lakes, NJ, USA).
SA-β-gal staining
We seeded mock-hEM and IL17RB-hEM in a 6-well plate (5.0 × 104 cells/well). IL17B (100ng/ml) was added to the culture medium and the cells were passaged repeatedly. After reaching the target number of passages, cells were stained according to the protocol of the Cellular Senescence Assay Kit (#CBA-230, CELL BIOLABS, INC., San Diego, CA, USA). Senescent cells were identified based on blue cytoplasmic staining under bright field microscopy (BZ-X700, Keyence): percentages of senescent cells per unit area (number of senescent cells/total cell count) was measured and compared with control conditions.
Apoptosis assay
Following organoid seeding and culture in a 48-well plate, we removed the ExM and washed the organoids. We added Cell Recovery Solution to remove the Matrigel from the plate, and then dispersed the organoids via pipette agitation and trypsinized them using TrypLE Express. The cell suspension was stained according to the protocol of the MEBCYTO Apoptosis Kit (#4700, MBL, Tokyo, Japan) and analyzed using a BD FACSMelody Cell Sorter.
Immunocytochemistry
We seeded the cells in collagen-coated eight-well chamber slides (3.0 × 104 cells/well), incubated them for 24 h at 37 °C under 5% CO2, and then incubated them for another 24 h in the presence or absence of IL17B (100 ng/ml). We fixed the cultures in 4% paraformaldehyde (#09154-85, Nacalai Tesque)/PBS, permeabilized them with 0.25% Triton X/PBS for 15 min at room temperature, and then blocked them with Protein Block Serum-Free (#X0909, Dako North America, Carpinteria, CA, USA) for 10 min at room temperature, with each step preceded by washing the sample three times with PBS. Next, we reacted the cells with primary antibody overnight at 4°C (anti-NF-κB p65, 1:100, #sc-372, Santa Cruz Biotechnology, Dallas, TX, USA), tripled-washed them in PBS, reacted them with secondary antibody for 30 min at room temperature out of direct light (Alexa Fluor 488 donkey anti-rabbit IgG(H + L), 1:500, #A21206, Invitrogen/Thermo Fisher Scientific), and triple-washed them in PBS again. We treated the reacted cells with ProLong Gold antifade reagent with DAPI (#P36935, Invitrogen/Thermo Fisher Scientific), covered them with a glass coverslip, and imaged the resulting fluorescence using a C2 confocal microscope (Nikon, Tokyo, Japan). We evaluated the intensity of the fluorescence in the images thus obtained using ImageJ software [41].
Immunofluorescence
We analyzed specimens of normal human endometrial tissue using immunofluorescence. These experiments were approved by the institutional review board of Kyushu University (approval no. 622-00). First, we prepared 4 μm paraffin sections, allowed them to air dry, and then deparaffinized them. We retrieved the antigens via microwave heating in 0.1% sodium azide at pH 9.0. We allowed them to cool for 20 min at room temperature and then blocked them for 10 min using Protein Block Serum-Free (#X0909, Dako North America). The samples were then allowed to react overnight at 4 °C with the following primary antibodies: CD68 monoclonal antibody (1:100, #76,437, Cell Signaling Technology), IL1β Mouse mAb (1:100, #12242S, Cell Signaling Technology), and anti-IL17B (1:100, #NBP2-11672, Novus Biologicals, Englewood, CO, USA). After washing them with PBS, we incubated them for 30 min at room temperature with the following secondary antibodies: Alexa Fluor 488 donkey anti-rabbit IgG(H + L) and Alexa Fluor 647 goat anti-mouse IgG(H + L) (1:1000 each, #A21206 and #A21236, Invitrogen/Thermo Fisher Scientific). We washed the reacted tissue with PBS, treated it with ProLong Gold antifade reagent with DAPI (#P36935, Invitrogen/Thermo Fisher Scientific), covered it with a glass coverslip, and imaged the fluorescence using a C2 confocal microscope (Nikon).
Monocyte separation
We used peripheral blood collected from laboratory volunteers using a Lymphoprep Tube (CosmoBio, Carlsbad, CA, USA) as a source of mononuclear cells. We isolated classic monocytes (CD14+/CD16−) from samples using the EasySep Human Monocyte Isolation kit according to the manufacturer’s protocol (#ST-19,359, Stemcell Technologies, Vancouver, BC, Canada). Once isolated, we cultured these cells in RPMI 1640 (#30264-56, Nacalai Tesque) supplemented with 50 ng/ml M-CSF (#300 − 25, PeproTech) to induce them to differentiate into macrophages. On day 6 of the culture, we activated the macrophages by adding lipopolysaccharide (LPS) and Pam3CSK4 (both 1 µg/ml; #L2630 and #P1585, Sigma–Aldrich). On day 7, we collected them for RNA extraction and, to confirm successful differentiation into macrophages, we trypsinized some cells and collected them from the dish, washed them, suspended them in FACS Buffer (PBS containing 2% FBS; 1.0 × 106 cells/sample), and blocked them for 15 min using FcR Blocking Reagent (#130-059-901, Miltenyi Biotec, North Rhine-Westphalia, Germany). We then washed them, suspended them in 100 µl FACS Buffer, and stained them for 15 min with CD68 monoclonal antibody (#76,437, Cell Signaling Technology) and FITC anti-human CD80 (#305,206, BioLegend, San Diego, CA, USA). Next, we washed them with FACS Buffer and then stained them for 15 min with PE Donkey anti-rabbit IgG (#406,421, BioLegend). Finally, we analyzed the cells using a BD FACSMelody Cell Sorter.
Time-lapse photography
We disaggregated endometrial tissue specimens donated by hysterectomy patients using the method described above. Each sample (1.0 × 107 cells) was washed, suspended in FACS Buffer, and blocked with anti-FcR mAb for 15 min. Next, we suspended it in 100 µl FACS Buffer and stained it for 15 min with FITC anti-human CD9 (#312,104 BioLegend), PE/Cyanine7 anti-human CD10 (#312,214, BioLegend), and APC anti-human IL17RB (#FAB 1207 A, R&D Systems, Minneapolis, MN, USA), after which we removed dead cells using propidium iodide. We then sorted the cells into IL17RB-positive and IL17RB-negative groups using the BD FACSMelody Cell Sorter. We seeded the two groups separately in 48-well plates at a density of 4.5 × 103 cells per 5 µl Matrigel and cultured them in ExM (250 µl/well) containing either IL17B only (100 ng/ml), SP600125 only (5 µM), or both. We grew these cultures at 37 °C under 5% CO2 for 7 d, during which we recorded them using time-lapse photography under a fluorescence microscope (BZ-X800, Keyence). We quantified the cultures in terms of organoid count (i.e., organoids ≥ 20 μm in diameter) per unit volume in Z-stacked images using a BZ-X700 microscope and BZ-X analysis software (BZ-X700 Analyzer, Keyence).
Statistical analysis
We performed all statistical analyses under advice from a professional clinical statistician, using GraphPad Prism 7 software on Windows 7.02 (GraphPad Software, San Diego, CA, USA). We used Student’s t-tests for comparisons between two independent groups and Dunnett’s multiple comparison test for multiple comparisons. Results data are presented with standard deviations; P < 0.05 was considered statistically significant. All analyses included post hoc tests.




留言 (0)