Study design
We used individual-level data from UK Biobank [17] and summary-level data from FinnGen [18] and Biobank Japan [19] for gallstone disease. We selected genetic mimics of plasma LDL-cholesterol lowering therapies from genes encoding the molecular target of each therapy. We conducted drug-target MR analyses to assess the associations of genetic mimics of each therapy with gallstone disease risk. We performed colocalization analyses to examine whether any associations found were driven by a shared causal variant between exposure and outcome or were confounded by linkage disequilibrium [20].
We extracted genetic predictors for plasma LDL-cholesterol from across the genome. We conducted clustered MR analyses to identify distinct clusters of genetic variants having similar causal estimates for plasma LDL-cholesterol on gallstone disease risk [21]. We performed pathway analyses to investigate biological pathways relating to each cluster. A summary of the study design is shown in eFig.1 (Online resource).
Data sources
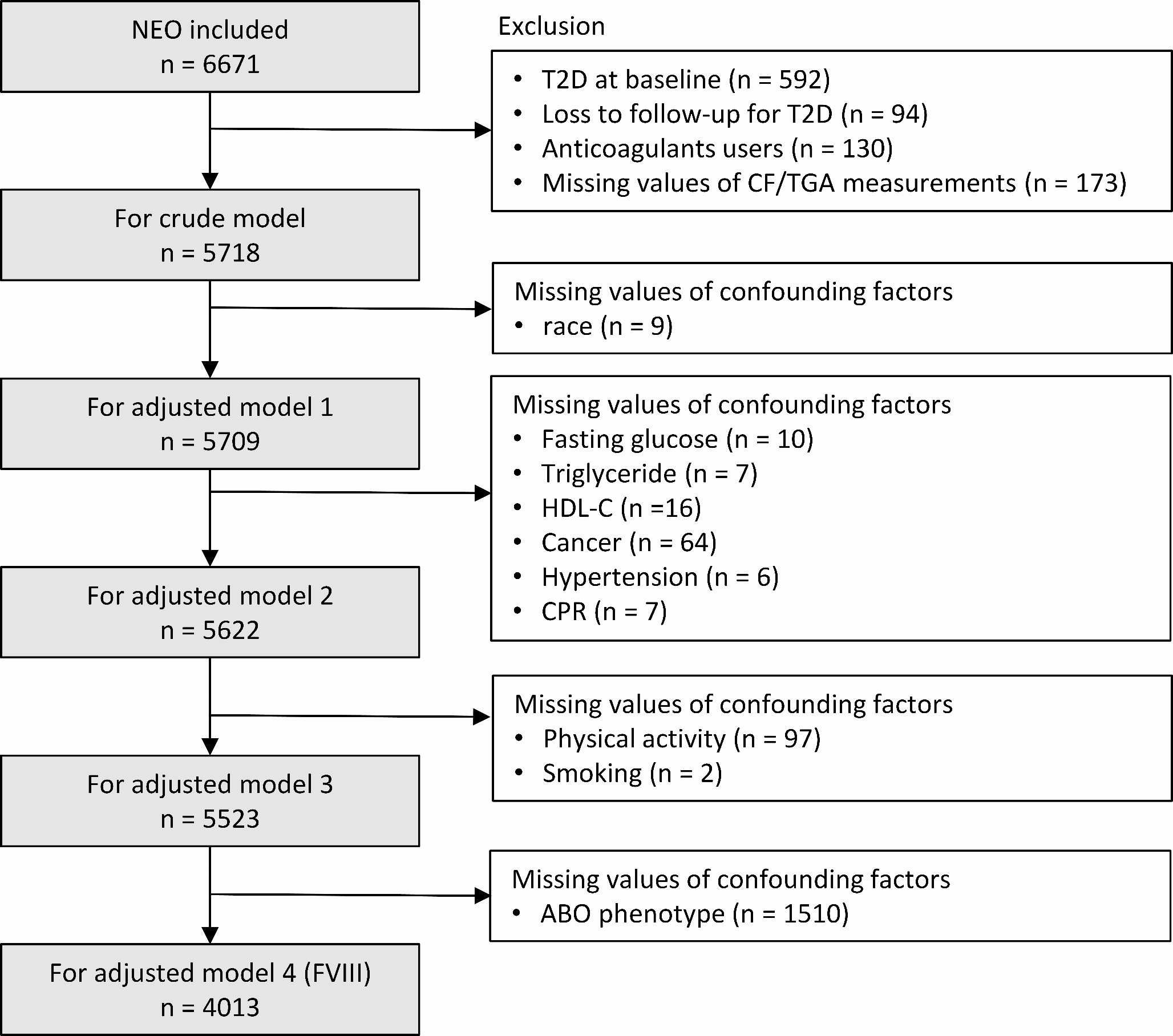
The UK Biobank recruited approximately 500,000 people (intended age 40–69 years, 94% self-reported European ancestry) between 2006 and 2010 from across the United Kingdom [17]. Individual-level data used were under application 98032 (October 2021 updated). Cases were defined based on self-reported history of gallstones, International Classification of Diseases (ICD)-9 and ICD-10 codes related to gallstones, and medical treatment of gallstones (Online resource: eTable 1), as previously [22]. Both prevalent and incident cases were included. Controls were individuals without gallstone-related disease or treatment. Individuals who underwent cholecystectomy due to an alternative pathology (e.g., neoplasm) were excluded. We included 367,289 unrelated individuals of European ancestry (30,547 gallstone disease cases and 336,742 controls) with genomic data passing quality control as described previously [23]. We used logistic regression to obtain sex-combined and sex-specific genetic associations with gallstone disease.
Summary-level data from FinnGen (R8 release) included 34,461 gallstone cases and 301,383 controls (mean age 52 years, 55.7% women) [18]. Cases were defined based on ICD-8, 9, 10 codes [18]. Summary-level data from Biobank Japan included 9,305 gallstone cases and 168,253 controls (mean age 63 years, 46.3% women) [19]. Cases were defined based on ICD-10 codes [19]. Definitive codes of gallstone disease are provided in eTable 1 (Online resource).
Genetic instruments
We selected genetic instruments based on their associations with plasma LDL-cholesterol obtained from ancestry-specific summary-level data from the Global Lipids Genetics Consortium (GLGC) (1,231,289/82,587 people of European/East Asian ancestry) [24], but not based on functional information. We selected genetic mimics of plasma LDL-cholesterol lowering therapies from genes encoding the molecular targets of each therapy (i.e., HMGCR for statins, PCSK9 for proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, NPC1L1 for ezetimibe, ACLY for ATP citrate lyase inhibitors, LDLR for targeting LDL receptors, and APOB for targeting apolipoprotein B (apoB)), as previously [25, 26]. We used genetic mimics of targeting ABCG5/8 from ABCG5/8 as a positive control exposure, because ABCG5/8 is a well-established lithogenic gene [2]. We included all variants within 100 kb on either side of each target gene that were in low linkage disequilibrium (r2 < 0.1) and were genome-wide significantly (p value < 5 × 10− 8) associated with plasma LDL-cholesterol. We used a less stringent cut-off for linkage disequilibrium (r2 < 0.1) to obtain more variants in each gene region to increase the power [27]. We also extracted genetic predictors for plasma LDL-cholesterol from across the genome that were uncorrelated (r2 < 0.001) and genome-wide significantly (p value < 5 × 10− 8) associated with plasma LDL-cholesterol. Estimates were expressed in 1-standard deviation (around 0.87 mmol/L) reduction in plasma LDL-cholesterol.
We used the F-statistic to assess instrument strength, approximated by the square of each SNP-exposure association divided by the square of its standard error [28]. We used PhenoScanner, a database of genotype-phenotype associations [29, 30], to check whether SNPs were genome-wide significantly (p value < 5 × 10− 8) associated with common confounders (i.e., socioeconomic status, smoking, alcohol drinking and physical activity). We included these SNPs in the main analysis, and excluded them in the sensitivity analysis. We also used positive control outcomes, i.e., CAD from Coronary ARtery DIsease Genome wide Replication and Meta-analysis plus The Coronary Artery Disease Genetics (CARDIoGRAMplusC4D) Consortium (60,801 cases and 123,504 controls) [31] for people of European ancestry and myocardial infarction (MI) from Biobank Japan (14,992 cases and 146,214 controls) [19] for East Asians. A summary of genome-wide association studies (GWAS) used is provided in eTable 2 (Online resource).
MR analysis
We aligned SNPs based on alleles and allele frequencies. We used proxy SNPs (r2 ≥ 0.8), where possible, when SNPs were not available in the outcome GWAS. We used the 1000 Genomes reference panel to obtain linkage disequilibrium. We selected a European population for proxy SNPs in the UK Biobank and FinnGen, and an East Asian population for proxy SNPs in Biobank Japan. We calculated MR estimates by meta-analyzing Wald estimates (the ratio of the genetic association with outcome to the genetic association with exposure) using inverse variance weighting (IVW) with fixed effects for three SNPs or fewer and random effects for four SNPs or more [32]. To assess the robustness of the IVW estimates, we conducted sensitivity analyses using weighted median [33] and MR Egger [34]. We used the MR Egger intercept to assess directional pleiotropy [34]. For uncorrelated SNPs (r2 < 0.001), we used IVW [32] and MR Egger [34] assuming independent genetic instruments. For SNPs in low linkage disequilibrium (r2 < 0.1), we used IVW [27] and MR Egger [35] extended to account for correlations between genetic instruments by fitting a generalized weighted linear regression model using a weighting matrix.
We meta-analyzed MR estimates from the three biobanks using a fixed-effects model unless the Q-statistic suggested heterogeneity when we used a random-effects model. We assessed differences by sex using a two-sided z-test [36].
Colocalization analysis
We performed colocalization analyses in a Bayesian framework to assess whether any associations found in drug-target MR were driven by a shared causal variant between plasma LDL-cholesterol and gallstone disease or were confounded by linkage disequilibrium [20]. This method uses Approximate Bayes Factor computations to assess the posterior probability of several hypotheses [20]. H0, no association with either trait; H1, association with plasma LDL-cholesterol only; H2, association with gallstone disease only; H3, associations of two independent variants and one for each trait; H4, associations of one shared variant with both traits [20]. A probability of H4 larger than 0.80 provides evidence for colocalization [20]. We included variants (minor allele frequency > 0.1%) in or near (+/-100 kb) the target gene where any associations were identified. We set the prior probabilities as recommended, i.e., 1.0e-4 for a variant associated with plasma LDL-cholesterol, 1.0e-4 for a variant associated with gallstone disease, and 1.0e-5 for a variant associated with both traits [20]. We also calculated the posterior probability for a shared variant associated with both traits conditional on the presence of a variant associated with gallstone disease, as the power to detect colocalization is low when the variants are not strongly associated with the outcome [37].
Clustered MR
We used the MR-Clust method to identify distinct clusters of genetic variants having similar causal estimates for plasma LDL-cholesterol on gallstone disease, which might reflect distinct biological pathways [21]. The MR-Clust accounts for differential uncertainty in the causal estimates, and includes a null cluster where SNP-specific estimates are centred around zero and a junk cluster where SNP-specific estimates are highly dispersed and are considered outliers [21]. The presence of null and junk clusters requires substantial evidence of similarity to define a cluster, which avoids the detection of spurious clusters [21]. We only included variants with inclusion probability > 0.80 in each cluster, and only reported a cluster if at least four variants satisfy this criterion, as recommended [21].
Pathway analysis
We performed pathway analysis to examine biological pathways relating to each variant cluster using the Functional Mapping and Annotation (FUMA) platform, which includes SNP2GENE and GENE2FUNC functions [38]. We first applied the SNP2GENE function to map cluster-specific genetic variants to genes, where we used 100-kb positional mapping and expression quantitative trait locus (eQTL) mapping based on GTEx v8 [38]. We then used the GENE2FUNC function to associate the mapped genes with biological pathways defined by KEGG and Reactome database [38].
All statistical analyses were conducted using R version 4.2.1 and the packages “ieugwasr”, “TwoSampleMR”, “MendelianRandomization”, “metafor”, “coloc”, and “mrclust”.






留言 (0)