
記住我
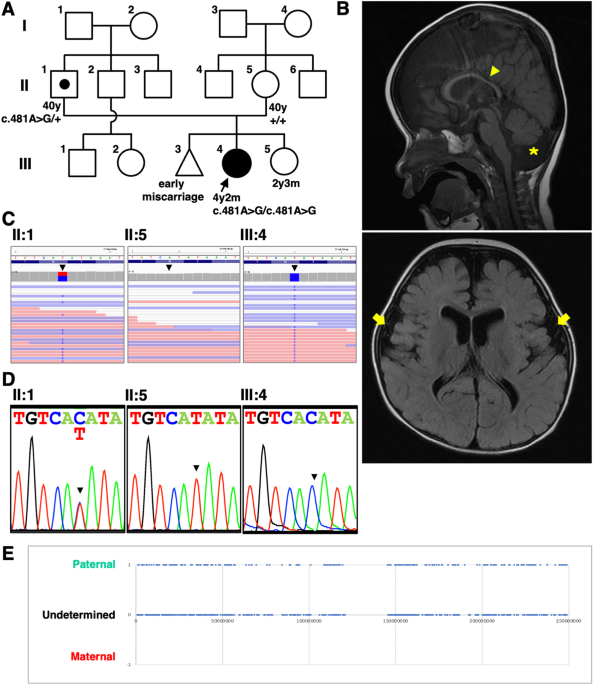
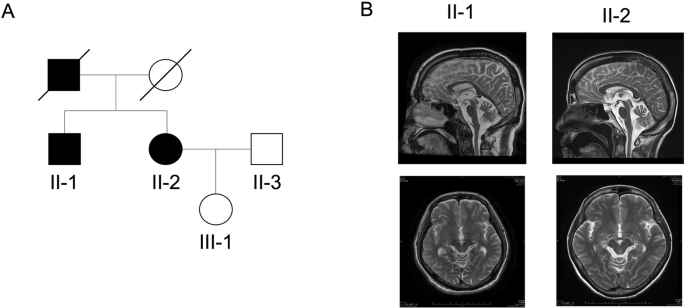
The female proband (III:4) was the second child of her healthy, nonconsanguineous Japanese parents (II:1 and II:5) (Fig. 1A). The first child was an early miscarriage of unknown cause. The third child was a girl healthy at 2 years and 3 months. There was no other family history to report. The patient was born at 40 weeks and 4 days of gestation. Her weight at birth was 3112 g (+0.1 SD), length was 49.0 cm (−0.4 SD), and head circumstance was 36.5 cm (+2.4 SD) (Supplementary Table S1). Although she showed no abnormality at birth, she was followed up due to a failure to thrive that became noticeable as generalized hypotonia and developmental delays (Table 1). Brain magnetic resonance imaging (MRI) showed cerebellar atrophy, decreased white matter, thin corpus callosum, wide opening of the Sylvian fissure, and larger ventricles in the brain at 1 year and 6 months of age (Fig. 1B). Although she passed the newborn hearing screening test, she was found to have bilateral moderate hearing loss at 1 year and 4 months. Treatment with tympanostomy tubes for otitis media was not effective for her hearing impairment, therefore she began wearing her hearing aids at age 2. A computed tomography (CT) scan revealed findings that were suspicious of bilateral ossicular malformations. Her growth impairment still existed at 4 years and 1 month: her weight was 12.1 kg (−2.1 SD), height was 92.8 cm (−1.9 SD) and head circumstance was 47.8 cm (−1.1 SD). She started walking at 2 years and 7 months and speaking at 3 years and 2 months. Her developmental quotient using the Kyoto Scale of Psychological Development 2020 was 50 at the age of 3 years and 6 months. She spoke only a few words but willingly tried to use nonverbal communication at the age of 4 years and 3 months. She began treatment with antiepileptic drugs at the age of 4 years to control recurrent seizures with a high fever. Neither ataxia, spasticity, nor involuntary movements had been presented during the follow-up period. These symptoms are not contradictory to those reported as neurodevelopmental disorders with hypotonia and cerebellar atrophy, with or without seizures, which are caused by genetic PIGK deficiencies [4, 5].
Fig. 1
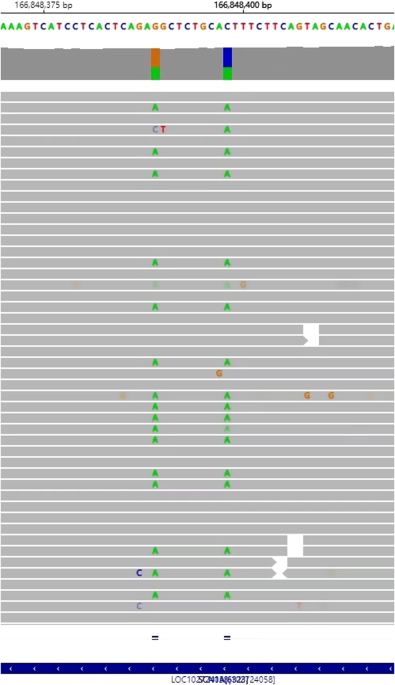
Clinical features and genetic analysis. A Pedigree of the family. The proband girl (III:4) was the second child of nonconsanguineous parents (II:1 and II:5). B Brain MRI of the patient at 1 year 6 months old. Cerebellar atrophy (asterisk), decreased white matter, thin corpus callosum (arrowhead), wide opening of the Sylvian fissures (arrows), and larger ventricles in brain were observed. C Integrative Genomics Viewer Images. The amino acid residues of PIGK are shown at the top and read counts are at left side of coverage track. Position of the candidate variant was indicated with black triangle. The c.481A > G missense variant of PIGK was homozygous in the proband (III:4) and heterozygous in the father (II:1). The mother had reference homozygous at c.481 (II:5). The coverage at the variant was comparative (proband: 50; father: 70; mother: 55). D Electropherogram of Sanger sequencing analysis. Heterozygous c.481A > G in the father (II:1); reference nucleotide c.481A in the mother (II:5); Homozygous c.481A > G in the proband (III:4). E Zygosity mapping using the WES data. For homozygous sequences of patients which passed data quality, the origin was estimated from the sequence data of the parents. The position of nucleotides estimated to have been inherited from the father was plotted as 1, the position of nucleotides estimated to have been inherited from the mother as -1, and the position of nucleotides that could still be inherited from either parent as 0. The horizontal axis indicates chromosome position
Table 1 Clinical features and PIGK variants in the present case and reported cases in literaturesWhole-exome sequencing analysisAfter obtaining informed consent from the patient’s parents, genomic DNAs were extracted from the peripheral blood in the family and trio-based whole-exome sequencing (WES) and filtering analysis were performed as previously described [18]. In brief, the sequence library was prepared using a Human All Exon V6 Kit (Agilent Technology, Santa Clara, CA, USA) and sequenced using NovaSeq (Illumina, CA, USA) with 150-bp paired-end reads. Sequence reads were aligned to GRCh38 and annotated using CompStor NOVOS and CompStor Insight (OmniTier, CA, USA). First, high-quality variants with allele frequencies greater than 0.01 in gnomAD, 14KJPN (jMORP), and our in-house exome variant data were removed. Next, the variants were narrowed down based on the assumed modes of inheritance, such as autosomal dominant, autosomal recessive, and X-linked inheritance. After filtration following this procedure, four variants, three homozygous variants, and one de novo variant were segregated (Supplementary Table S2). We further curated these variants by searching OMIM and PubMed databases, and prediction tools, CADD, Polyphen-2, SIFT, and MutationTaster. The nucleotide sequence of the variant region was confirmed by Sanger sequencing (ABI3130, Life Technologies, CA, USA). Genomic DNA was amplified using Ex-Taq (TAKARA, Shiga, Japan) with the forward primer; 5′-TAACTGGGAGGATCCCACCTAGT-3′ and reverse primer; 5′-CAGTTGGTAATCTTCATGTACAGC-3′. Electropherograms of the sequencing result were obtained using reverse primer. Zygosity analysis on chromosome 1 using the WES data was performed previously reported using an in-house program [19]. In brief, homozygous nucleotides of the patients were selected and their origin was estimated by reference to the parental genotypes. Nucleotide positions that could have been inherited from the father were plotted as 1, those that could have been inherited from the mother as −1, and those that could have been inherited from either parent as 0.
In silico three-dimensional structural analysis of the PIGK proteinData on the cryo-EM structure of the human GPI transamidase complex (PDB code: 7WLD) [20, 21] were imported into the Molecular Operating Environment (MOE). The methionine at position 161 was replaced with valine or leucine, and the three-dimensional views around the amino acids were calculated.
Phylogenetic analysisAmino acid sequences of vertebrate PIGK proteins were obtained from the NCBI database. The accession numbers are as follows; Human: NP_005473; Rhesus monkey: NP_001247873; Mouse: NP_079938; Chicken: NP_001026449; Common lizard: XP_034977403; Frog (Xenopus tropicalis): NP_001072709; and Zebrafish: NP_001002149. Genetyx-Mac Ver. 22 (Genetyx Corporation, Tokyo, Japan) was used to construct a protein alignment.
CHO cell analysisThe empty pME or pTK vectors or human PIGK (wild-type or p.Met161Val) expression plasmids were electroporated to 107 cells of PIGK-deficient Chinese hamster ovary (CHO) cells (clone 10.2.2) [22]. Two days after electroporation, the 2 × 104 cells were analyzed in a single run by fluorescence-activated cell sorting (FACS) using a flow cytometer (MACSQuant Analyzer; Miltenyi Biotec, Bergisch Gladbach, Germany) with FlowJo software (Tommy Digital, Tokyo, Japan) as described previously [5]. The following antibodies were used for cell labeling: mouse anti-human CD59 (clone 5H8); anti-human decay accelerating factor (DAF) (clone IA10, BD Biosciences); mouse anti-hamster urokinase-type plasminogen activator receptor (uPAR) (clone 5D6); and phycoerythrin-conjugated anti-mouse IgG antibody. Whole-cell lysates of 106 CHO cells were subjected to SDS-PAGE and Western blotting. Expression of PIGK proteins was detected by goat anti-GST polyclonal antibody (Sigma-Aldrich, St. Louis, USA), followed by HRP-conjugated anti-goat IgG. For the loading control, GAPDH was detected by mouse anti-GAPDH (AM4300, Invitrogen, Waltham, MA, USA), followed by HRP-conjugated anti-mouse IgG.
AnimalsZebrafish (Danio rerio) were reared and maintained in 1.7 L tanks in a recirculating system (Meito System, Nagoya, Japan) at 28.5 °C under a 14 h light and 10 h dark photoperiod. Larvae were fed paramecia and Gemma Micro ZF 75 (Skretting, Westbrook, ME, USA) twice daily from 5 to 30 days postfertilization (dpf). Juvenile fish were fed brine shrimp (Tokai Guppy, Okazaki, Japan) and Gemma Micro ZF 75 twice daily from 30 to 90 dpf. Adult fish were fed brine shrimp and Otohime B2 (Marubeni Nissin Feed, Tokyo, Japan) twice daily after 90 dpf. We obtained a zebrafish pigktt261 mutant allele from the Zebrafish International Resource Center. This pigktt261 allele carries a T to C mutation in the first exon of the pigk gene, disrupting the first methionine codon [23]. The Tg(zCREST2-hsp70:GFP) transgenic fish [24] used to visualize sensory Rohon–Beard (RB) neurons in electrophysiology were provided through the National BioResource Project of Japan.
Zebrafish analysesFor rescue experiments, full-length cDNAs encoding human PIGK were cloned into the expression vector pCS2+ using the In-Fusion HD Cloning Kit (Clontech, Mountain View, CA, USA) according to the manufacturer’s manual. The p.Met161Val, p.Gln33TER, p.Ser53Phe, p.Ala87Val, p.Tyr160Ser, p.Asp204His, and p.Cys275Arg mutations were introduced into the human PIGK expression construct by site-directed mutagenesis, as described previously [25]. Primers used for cloning and mutagenesis are listed in Supplementary Table S3. Expression constructs were used to generate human PIGKWT, PIGKM161V, PIGKQ33X, PIGKS53F, PIGKA87V, PIGKY160S, PIGKD204H, and PIGKC275R mRNAs using the mMESSAGE mMACHINE SP6 Kit (Thermo Fisher Scientific, Waltham, MA, USA) as described previously [25]. Human PIGK mRNAs (100 pg) were injected into 1-cell-stage zebrafish embryos produced by crossing PIGKtt261 heterozygous fish. For genotyping, the genome was extracted from mRNA-injected embryos, as described previously [26]. The genomic pigk region surrounding the T to C mutation (ATG to ACG in the first methionine codon) was amplified by PCR and subjected to direct sequencing using an Applied Biosystems 3500 Genetic Analyzer (Thermo Fisher Scientific).
For the touch response, tactile stimulation was applied to the tails of zebrafish embryos at 48 h postfertilization (hpf). Touch responses were recorded at 200 frames per second using a high-speed camera HAS-220 (Ditect, Tokyo, Japan), as described previously [27]. Embryos that showed swimming following a touch were judged as normal, whereas embryos that did not respond to a touch were judged as unresponsive. Mutant embryos that showed the recovery of touch response displayed normal swimming locomotion in appearance.
For in situ hybridization, full-length cDNAs encoding zebrafish pigk were cloned into the expression vector pCS2+. Primers used for cloning and mutagenesis are listed in Supplementary Table S3. DIG-labeled riboprobes were synthesized and used for RNA labeling with NBT/BCIP (Roche, Basel, Switzerland) according to established procedures [28].
For electrophysiology, zebrafish embryos (48 hpf) carrying zCREST2-hsp70:GFP transgene [24] were used to visualize RB cells. The dissection protocols for in vivo patch recordings in RB neurons have been described previously [23, 29]. The resting membrane potentials of the patched RB cells (−58 ~ −68 mV) were comparable between wild-type and pigk mutant embryos.
For immunohistochemistry, skin-removed zebrafish embryos (48 hpf) were fixed in 4% paraformaldehyde and subjected to immunolabeling, as described previously [28]. The following primary antibodies were used: rabbit anti-voltage-gated sodium channel Pan (SP19, 1:500; Sigma-Aldrich) and mouse monoclonal anti-HuC/HuD (16A11, 1:500; Thermo Fisher Scientific). Alexa 488-conjugated anti-rabbit IgG and Alexa 568-conjugated anti-mouse IgG (1:500; Thermo Fisher Scientific) were used as the secondary antibodies. Fluorescence images were captured using a confocal microscope TCS SP5 (Leica Microsystems, Wetzlar, Germany). The Nav labeling at the cell body was observed in all wild-type and pigk mutant RB cells, but Nav labeling at axons was seen in 20% of wild-type RB cells but not at all in mutant RB cells.
留言 (0)