Blood samples
A total of 80 (52 males and 28 females; age 42 to 75 years old) newly diagnosed MM patients without co-morbidities, including other cancers, diabetes, cardiovascular disease, and neurological disease in our hospital from October 2017 to April 2019 were enrolled. MM diagnosis, staging, and risk status were in accordance with the National Comprehensive Cancer Network Guidelines (3rd Edition, 2017). A total of 65 healthy subjects (43 males and 22 females; age, 40 to 78 years) were enrolled as the control group during the same period. The procedures in this study followed were in accordance with the ethical standards of the responsible committee on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008, and was approved by The Third Xiangya Hospital ethics committee (NO.2017S124). All human participants have given written informed consent.
Cell culture and treatment
U266, RPMI8226 cells were purchased from the cell bank of Shanghai Institute of Biology (Shanghai, China), and were cultured in 1640 complete medium with 10% extracellular vesicle-depleted Fetal Bovine Serum (cat no. A2720801, Gibco, USA) at 37 °C, 5% CO2 incubator. For LRG1-blocking treatment, the cells were treated with 5 μg/ml of the anti-LRG1 antibody C4 (Santa Cruz Biotechnology, Inc. Dallas, Texas, USA) for 48 h and used for further analysis. For exosome treatment, the cells were treated with exosomes collected from 10 ml peripheral blood for 48 h and used for further analysis.
Platelet activation assay
Fasting venous blood was collected and anticoagulated with sodium citrate. Specimens were tested within 15 min after collection. 1 ml of whole blood samples were added with 1 μg human CD61-PERCP (Becton Dickinson Biosciences, cat: 340506), 1 μg CD62P PE (Becton Dickinson Biosciences, cat: 348107) for 15 min incubation. Then, the samples were fixed with ice-cold 1% paraformaldehyde and analyzed by Flow cytometry (FACSCalliber, BD Biosciences, San Diego, CA) for CD61, CD62P, and percent double positive.
Preparation of platelet-rich plasma (PRP)
About 10 ml peripheral blood was drawn from MM patients and healthy controls into 8% sodium citrate anticoagulation tubes. Whole peripheral blood was centrifuged at low-speed centrifugation of 200 × g for 10 min to obtain a PRP. PRP were centrifuged at 2000 × g for 10 min to obtain platelet-poor plasma (PPP) from supernatants, and platelets were purified by resuspension in modified calcium-free Tyrode buffer.
Determination of platelet aggregation rate
The platelet aggregation rate was measured in PRP by stimulating with ADP (10 μM, Sigma-Aldrich, St. Louis, MO, USA) and detected by turbidimetric method with aggregator (Beijing Pulisheng LBYNJ4, Beijing, China). The maximum platelet aggregation rate was recorded.
Detection of proliferation by 5-ethynyl-2’- deoxyuridine (EdU) fluorescent staining
RPMI8226 and U266 cells were treated with platelet-rich plasma or exosomes for 48 h. The EdU solution (50 μmol/L) was added and incubated with cells for 2 h, and the nuclear were stained with DAPI (2 μg/ml) for 5 min.
Detection of apoptosis by flow cytometry
After treatment with platelet-rich plasma or exosomes for 48 h, the RPMI8226 and U266 cells were obtained by centrifugation at 2000 × g for 5 min. A Annexin V-PE apoptosis detection kit (KeyGen Bio Tech, Nanjing, China) was used to measure the apoptotic cells according to the instructions. The results were analyzed by using BD FlowJo software (V10). The apoptotic cells included annexin V+ single positive cells plus annexin V+ PI+ positive cells.
Western blot
The tumor tissue or the treated cells were collected to extract proteins by RIPA Lysis Buffer (Beyotime, Shanghai, China). The proteins (30 μg) were separated with SDS gel and transferred to the PVDF membrane. After blocked with 5% BSA for 1 h, the membranes were incubated with the primary antibodies (Bcl2 (Rabbit monoclonal [E17], dilution, 1:1000), Bax (Rabbit monoclonal [E63], dilution, 1:1000), GAPDH (Rabbit monoclonal [EPR16891], dilution, 1:1000), LRG1(Rabbit monoclonal [EPR12362], dilution, 1:1000), CD63 (Rabbit monoclonal [EPR5702], dilution, 1:1000), E-cadherin (Rabbit monoclonal [EP700Y], dilution, 1:1000), N-cadherin (Rabbit monoclonal [EPR22397-264], dilution, 1:1000), Vimentin (Rabbit polyclonal (V9), dilution, 1:1000), VEGF (Rabbit monoclonal [EPR9699], dilution, 1:1000), MPO (Rabbit monoclonal [EPR20257], dilution, 1:1000), SAA1 (Rabbit monoclonal [D9H4L41], dilution, 1:1000), Transferrin (Rabbit monoclonal [EPR18240-29], dilution, 1:1000); from Abcam, UK. S100A8 (Rabbit monoclonal [E4F8V], dilution, 1:1000), S100A9 (Rabbit monoclonal [D3U8M], dilution, 1:1000), PCNA ((Rabbit monoclonal [13110], dilution, 1:1000) from Cell Signaling Technology, USA) overnight at 4 °C. After washed with TBST, the membranes were incubated with secondary antibodies (Goat Anti-Rabbit IgG H&L (HRP), dilution, 1:5000, cat no. ab205718, Abcam) for 1 h. The bands were obtained by ChemiDoc™ XRS system (Bio-Rad). ImageJ was used to quantify the bands.
Proteome analysis
The peripheral blood was obtained from three pairs of MM patients and healthy subjects matched by sex and age (Supplementary Table 1). Unlabeled quantitative protein spectrum was used to detect the expression of proteins in the samples. Protein identification was performed using Precursor Qvalue. Product ion peak area was used for protein quantification, and at least three product ions were selected for quantification; Students t-test was used for protein difference analysis; protein function analysis was based on GO, KEGG.
Extraction and identification of PRP exosomes
The PRP was obtained as abovementioned, and centrifuged at 150,000×g for 120 min. The supernatant was discarded, and then 100 μL of PBS was added to resuspend the exosome pellet. The exosomes were identified by transmission electron microscope and analyzed by Nanoparticle tracking analysis (NTA, NanoSight NS300, Malvern, Cambridge, United Kingdom). The expressions of exosome-related marker proteins CD63 and specific platelet marker CD41 were detected by flow cytometry.
Measurement of exosomal LRG1 levels
Exosomal LRG1 levels were quantified by Human LRG1 ELISA Kit (cat no. ab260066, Abcam, UK) according to the manufacturer’s protocol.
Fluorescently labeled exosomes
The cell membrane was stained with wheat germ agglutinin (cat no. W11261, Thermo Fisher, USA) according to the manufacturer’s protocol. PKH26 dye (2 μM, Solarbio, Beijing, China) was added and incubated with the platelet-derived exosome suspension for 20 min at room temperature, protecting it from light. PKH26-labeled exosomes were co-incubated with U266 and RPMI8226 cells for 24 h. The cells were washed twice with PBS and then resuspended in a culture medium; The exosome tracking was observed by confocal microscopy.
Plasmids construction and transfection
The full-length LRG1, OLFM1, OLFM2, OLFM3, and OLFM4, and their truncated fragments (LRG1Δ1: 13–310 aa; LRG1Δ2: 1–12, 264–310 aa; LRG1Δ3: 1–263 aa; OLFM4Δ1: 155-507 aa; OLFM4Δ2: 31–71, 245–507 aa; OLFM4Δ3: 31–234 aa) were directly synthesized by Songon Biotech Corporation (Shanghai, China), and were inserted into pCMV-N-HA (cat: D2733-1, Beyotime) or pCMV-N-Flag (cat: D2722-1, Beyotime, Shanghai, China) plasmids to construct the recombinant plasmids. The nucleotide sequences of all recombinant plasmids were verified by DNA sequencing (Songon Biotech Corporation, Shanghai, China)). For transfection, 20 nM plasmids or empty plasmids (control) that were mixed with Lip3000 were added into the U266 cells and incubated for 48 h. The transfected U266 cells were used for further analysis.
Immunoprecipitation (IP)
Forty-eight hours after transfection of the expression plasmid, cell lysis buffer (containing protease inhibitors (10 μl, cat no. 78425, Thermo Fisher, USA) was added to lyse U266 cells for 30 min, and the supernatant was collected after centrifugation at 12,000 × g for 30 min. Anti-HA antibody (10 μg, Rabbit polyclonal, cat no. ab9110, Abcam) or Anti-Flag antibody (10 μg, Rabbit polyclonal, cat no. ab205606, Abcam) and 50 μl protein A/G-beads (B23202, Protein A/G immunoprecipitation magnetic beads, Bimake, USA) were added to the lysate (1 mg) respectively and incubated overnight at 4 °C with slow shaking. The protein A/G-beads were collected and was washed four times with 0.5 ml lysis buffer and centrifuged at 3000 × g for 2 min. The proteins were collected to perform a Western blot.
Immunofluorescence staining
Forty-eight hours after transfection of each recombinant plasmid into cells, the cells were crawled, fixed with 4% paraformaldehyde, permeabilized with 0.1% triton, blocked with donkey serum for 1.5 h, and incubated with primary antibodies (E-cadherin (Rabbit monoclonal [EP700Y], dilution, 1:1000), N-cadherin (Rabbit monoclonal [EPR22397-264], dilution, 1:1000), Vimentin (Rabbit polyclonal, dilution, 1:1000), VEGF (Rabbit monoclonal [EPR9699], dilution, 1:1000); from Abcam, UK) overnight at 4 °C. PBS was used as negative control. Fluorescent secondary antibodies (AlexaFluor488 donkey anti-rabbit, cat no. A21206, green; Alexa Fluor 594 goat anti-rabbit, cat no. R37117, red; Thermo Fisher, USA) were incubated for 1 h at room temperature. DAPI were used to stain the nuclei. The images were captured under a fluorescence microscope (Leica, USA).
Endothelial cell tube formation assay
The Matrigel matrix (growth factor reduced) and pipette were pre-cooled overnight at 4 °C. About 60 μl of Matrigel matrix was injected into a 96-well plate and refrigerated again at 4 °C overnight. The plated 96-well plate was incubated at 37 °C for 30 min, the HUVECs cells were resuspended in medium, and 100 μl of HUVECs cells (3 × 105 cells/ml) was added to each well. About 100 μl of treated U266 and RPMI8226 culture supernatants were added to each well. The tube formation of HUVECs in each group was observed under an inverted microscope (Mateo TL, Leica, USA). ImageJ plugin Angiogenesis Analyzer was used to quantify the vessel formation, including branch points and capillary length.
NOD/SCID mouse subcutaneous transplantation model
NOD/SCID mice (4–5-week-old male, 18–20 g, n = 5/group) were purchased from SJA Laboratory Animal Co. LTD (Changsha, China). 1.0 × 106 U266 cells (100 μl) transfected with LRG1 or OLFM siRNA were subcutaneously injected into the mice. The tumor volume was measured every 2 days by investigators who was blinded to the group allocation during the experiment. The animal experiments were approved by the animal ethics committee of Central South University and strictly followed the institutional and national guide for the care and use of laboratory animals.
Immunohistochemistry
After tumor tissue was taken, the tissues were routinely fixed, embedded, and serially sectioned with a thickness of 4 μm and dewaxed with xylene. Gradient ethanol hydration treatment, blocking endogenous peroxidase with 3% hydrogen peroxide solution, washing with PBS buffer (pH = 7.2–7.4), and antigen retrieval with EDTA (pH = 8.0), and then incubated with primary antibody (mouse anti-human Ki67, 1:500) in a 37-degree water bath for 1 h, and add EnVision secondary antibody in a water bath for 30 min, rinse with PBS buffer, and stained with freshly prepared DAB solution. The reaction time was controlled under the microscope, the color development was stopped with tap water, the nuclei were counterstained with Mayer, and observed under the microscope.
TUNEL staining
The tumor tissue samples were embedded with paraffin and cut to a thickness of 4 μm. The cut sections were heated in an oven at 45 °C overnight. After deparaffinization and antigen retrieval, proteinase K was diluted to a solution of 20 μg/mL with 10 mmol/L Tris-hydrochloric acid (pH 7.4–7.8), and was used to treat the sample for 20 min at 37 °C. After being washed twice with phosphate buffer solution (PBS) for 5 min each, the samples were incubated with a TUNEL reaction mixture for 60 min at 37 °C. Nuclei were then counterstained with DAPI. Apoptotic cells positive for TUNEL staining were observed under a microscope.
Immunofluorescence analysis for CD34 and vascular endothelial growth factor (VEGF)
Immunofluorescence detection of CD34 and VEGF was performed on the excised tumor tissues. The slides were fixed in methanol at −20 °C for 5 min, placed in phosphate buffered solution containing 0.05% Tween-20 for 5 min, and then blocked with PBS containing 5% BSA for 1 h. After incubation with antibodies CD34 and VEGF (1:100, Abcam, UK) for 1 h, the slides were gently rinsed three times with PBST (3 min/time), and then incubated with secondary antibodies conjugated with AlexaFluor488 goat anti-mouse and AlexaFluor568 goat anti-rabbit IgG (IgG; H + L; 1:200, Invitrogen, USA) for 30 min. Images were captured using a confocal microscope (PerkinElmer, USA). ImageJ was used to quantify the density of fluorescence.
Statistics
Kaplan–Meier analysis was performed to generate overall survival (OS) curves. Comparisons between two groups of data were analyzed using Student’s t-test, and multiple sets of data were analyzed with one-way ANOVA; data were presented as the means ± SEM using GraphPad Prism 8.01. P values less than 0.05 indicates statistical significance.

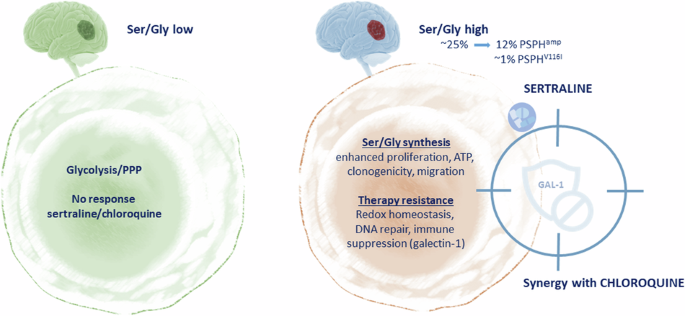







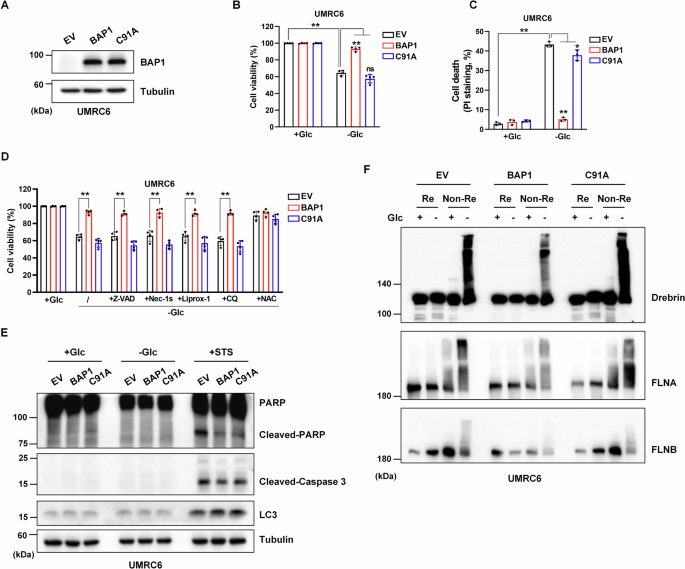
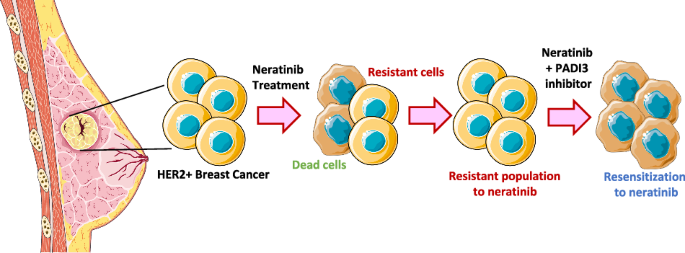
留言 (0)