Inclusion/exclusion criteria
In this RCT, we will enroll very and extremely premature and very and extremely low birth weight infants (birth weight < 1250 g and/or a gestational age < 30 weeks) who require insertion of an UVC for parenteral nutritional and/or drug administration because this represents the “standard population” of premature infants that will most commonly require prolonged central vascular access.
Intervention(s)
The “UVC—You Will See Study” is a pragmatic RCT comparing two different dwell times within the manufacturer’s recommendations. While the manufacturers’ specifications allow dwell times of up to 14 days, it is common clinical practice to remove UVCs in most NICUs within the first days of life. Moreover, data from our pilot RCT “UVC—You Will See Study” indicate that a dwell time > 10 days is associated with an increased rate of UVC-associated complications (unpublished data).
Outcome measures
We chose primary, key secondary, and secondary endpoints, as well as an exploratory endpoint since it is considered the “gold standard” in the assessment of catheter-related complications. Importantly, in doing so, we will also assess potential benefits associated with a longer dwell time (i.e., fewer painful invasive procedures for vascular access, less radiation exposure, fewer days of antibiotics, reduction in medical expenditures). BPD, NEC, FIP, IVH, PVL, and ROP comprise all major neonatal morbidities, and mortality will be assessed.
Primary efficacy endpoint
Number of catheter-related bloodstream infections and/or catheter-related thromboses/emboli and/or catheter-associated organ injuries including cardiac arrhythmias related to the use of UVC and/or peripherally inserted central catheters (PICC).
Key secondary endpoint(s)
Number of catheter-related bloodstream infections and/or catheter-related thromboses/emboli and/or catheter-associated organ injuries including cardiac arrhythmias related to the use of UVC.
Secondary endpoints
Number of painful procedures associated with insertion of UVC, PICC, and peripheral catheters; number of X‑rays for assessment of correct placement of UVC/PICC (radiation exposure); use of antibiotics with regard to suspected/proven CVC-associated (UVC/PICC) bloodstream infection; medical costs associated with the central venous catheters (UVC and PICC).
Exploratory endpoint
Correlation between length of dwell time and primary outcome parameter. Assessment of safety: Standardized clinical and ultrasonographic assessment as per study protocol; additional septic work-up as indicated as well as electrocardiography (ECG) examination in case of cardiac arrhythmia.
Methods against bias
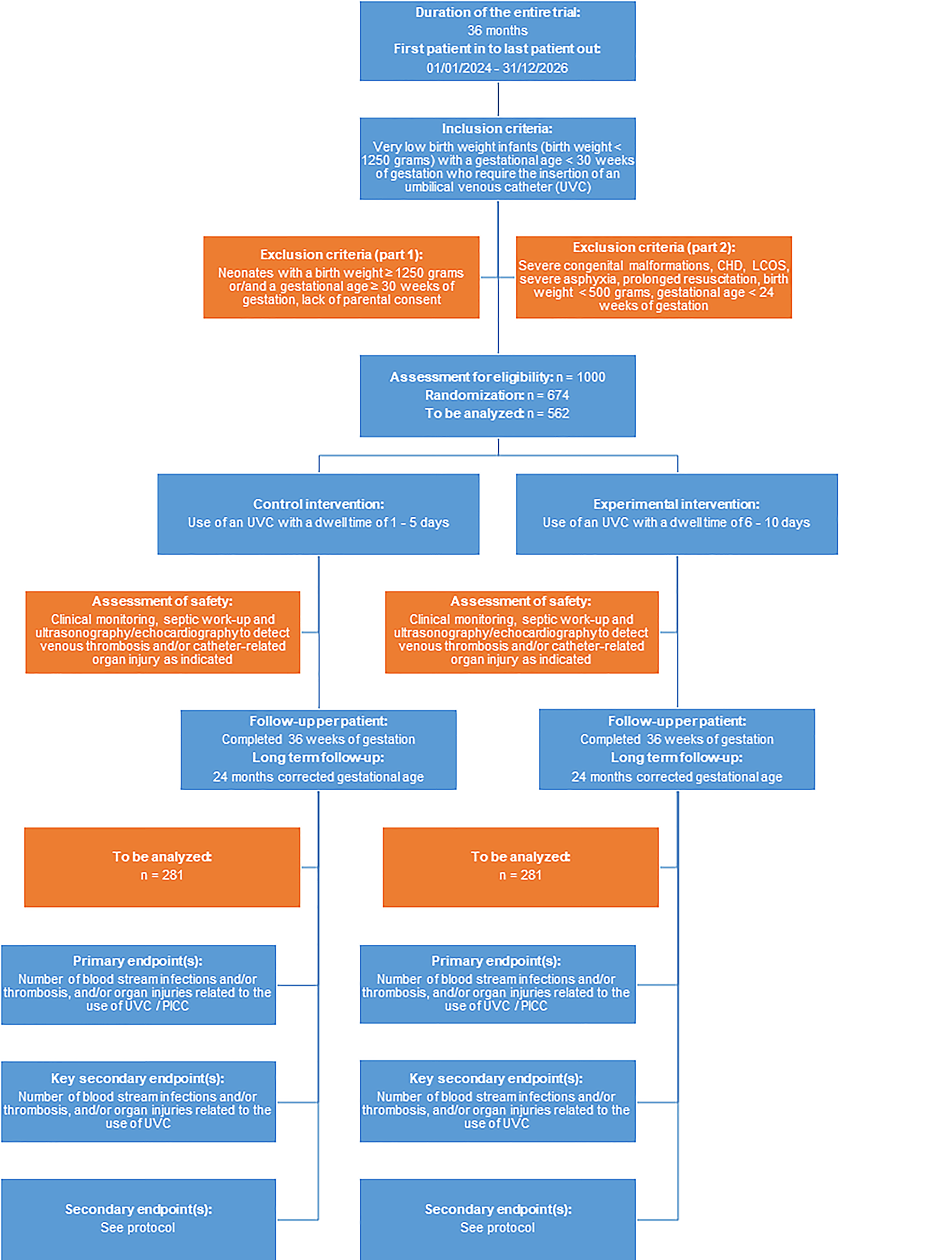
This trial will be conducted as a multicenter, active-controlled RCT. Neonates with a birth weight < 1250 g and/or a gestational age < 30 weeks who require UVC for parenteral nutrition and/or drug administration will be randomized to either a catheter dwell time of 1–5 days (standard therapy) vs. a dwell time of 6–10 days (interventional therapy). A randomization list will be generated by the Interdisciplinary Centre for Clinical Trials (IZKS) Mainz. The randomization ratio will be 1:1 using block randomization. A web-based randomization tool developed by IZKS Mainz will be used in this trial, allowing investigators to randomize patients via a secure web interface. While randomization is feasible, blinding of treating physicians is not possible due to the nature of the intervention. However, assessment of major outcome parameters (e.g., number of painful procedures, radiation exposure, use of antibiotics, medical costs) will be assessed by an independent researcher blinded to dwell times.
Proposed sample size/power calculations
For the sample size calculation—based on an extensive literature review, a current survey of participating centers, and results from our pilot study—we assume an event rate of 30% in the control group. The non-inferiority bound was fixed to 10%. The sample size was planned with a one-sided level of significance of 5% and a power of 80% using a two-sample t‑test. Non-inferiority will be concluded if the one-sided 95% confidence interval of the treatment difference is located completely below the non-inferiority bound. The sample size amounts to 562 (= 2 × 281) evaluable patients. The primary analysis is the per protocol population consisting of all randomized patients complying sufficiently with the study protocol. When assuming 20% of the randomized patients will not be part of the per-protocol population, 674 patients will have to be randomized (Fig. 1). The sample size was calculated with SAS version 9.4 (Cary, NC, USA).
Feasibility of recruitment
All participating centers are level-III NICUs with extensive expertise in the management of VLBW and ELBW infants. The participating centers have been actively involved in a number of large, multicenter neonatal trials, and have repeatedly demonstrated their competency in successfully recruiting adequate numbers of patients in a number of different trials (e.g., 12/14 centers actively participate in the NeoVitaA trial), and all 14 centers have provided full commitment to study participation and adequate patient enrollment. In our pilot single-center RCT “UVC—You Will See,” we enrolled 64 patients within an 18-month period. Thus, the recruitment of 562 infants by 14 level-III NICUs over a 3-year period is feasible. Additional centers will be contacted for potential study participation if required.
Statistical analysis
The primary endpoint will be analyzed within a logistic regression model with treatment as the fixed factor and center as covariate. Non-inferiority will be concluded if the one-sided 95% confidence interval of the absolute risk difference of the experimental intervention and the control intervention is completely located below the non-inferiority bound of 10%. The primary analysis population is the per-protocol population consisting of all randomized patients who sufficiently comply with the protocol, i.e., all subjects without violation of any exclusion or inclusion criteria and with an UVC at least 1 or 6 days respectively. For the experimental intervention, we expect a UVC dwell time of 6–10 days in 90% of the patients. The population is chosen very liberally so as not to introduce any bias, e.g., by excluding informative dropouts. The results in the ITT population will be considered as equally important. Since the primary endpoint is composite, the analysis will be repeated for every single component. Several sensitivity analyses will be performed. Additional parameters like birth weight, gestational age, hematocrit, or gender will be included in the analysis. Descriptive statistics will be displayed whenever appropriate.
The key secondary endpoint will be analyzed by the same model as the primary analysis. However, the objective is to show superiority of the experimental treatment versus the control treatment. Other secondary endpoints will be analyzed by a negative binomial regression model. Although they are considered exploratory, they will be interpreted at a significance level of α = 5%.

留言 (0)