
記住我





This study will be conducted in a 27-bed adult ICU at a large urban Australian tertiary referral hospital specialising in cardiothoracic surgery and medicine. The ICU originally comprised 21 open-plan bedspaces and six isolation beds in three ‘pods’ of nine beds each, surrounding a central nursing station. Two of the windowless open-plan bedspaces were modified and will serve as the intervention beds; the remaining 25 will function as control beds. The unit admits approximately 1800 patients annually.
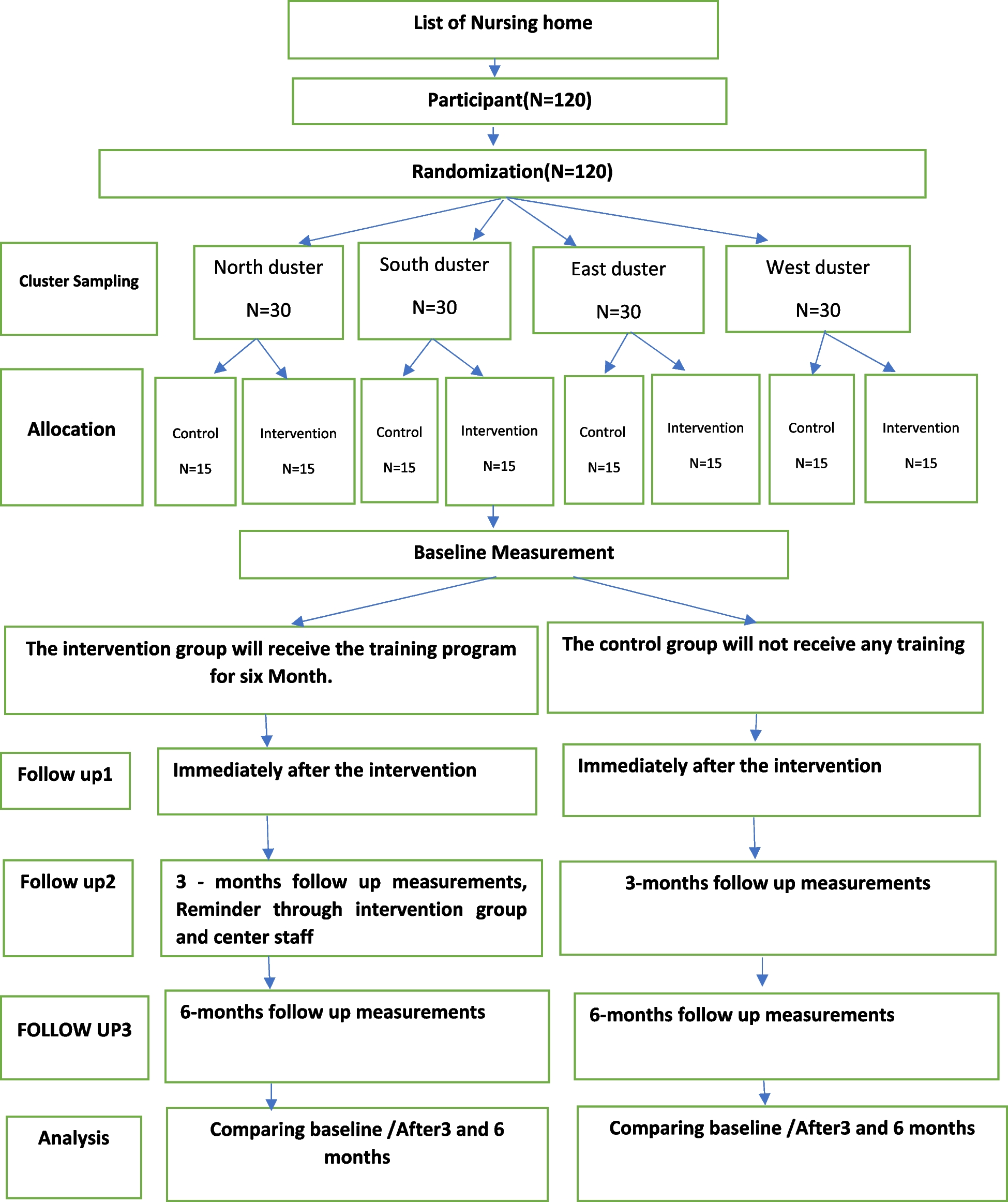
Study design and planThis is a prospective multi-component, mixed methods study including a randomised controlled trial (RCT) (Fig. 1). The study followed the SPIRIT reporting guidelines (Supplementary File) [34].
Fig. 1
Randomisation, recruitment, enrolment, and study plan
Study components encompass:
1.An objective environmental assessment
2.A description of the ICU environment from the perspective of patients, families, and staff
3.Sleep and circadian rhythm investigations
4.Delirium measurements
5.Assessment of medium-term patient outcomes 6 months after discharge from ICU
6.A health economic evaluation
Data pertinent to each component will be collected and analysed using methods appropriate to type and source of data and analysed separately before being synthesised in a summative assessment.
RCT componentThe primary study assessing patient outcomes (study components 3–6 described below) is an RCT, in which the two upgraded bedspaces are defined as intervention, while the 25 ‘conventional’ bedspaces serve as control. Eligibility screening, consent, and randomised allocation is a two-step process. All patients admitted to ICU are eligible to be randomised 1:1, except those deemed unsuitable for admission to the intervention bedspaces:
Patients requiring extracorporeal membrane oxygenation, intra-aortic balloon pump, or dialysis
Patients admitted to ICU immediately following cardiac surgery, including transplantation
Patients will be randomly assigned to bedspaces as follows:
1.A random sequence of allocations to intervention and control bedspaces was established by the research coordinator and sealed in numbered, opaque envelopes. ICU floor coordinators and clinical personnel are blinded to this sequence until revealed at randomisation.
2.Randomisation is completed by the ICU floor coordinator/admitting ICU doctor when:
At least one intervention and one control bedspace are available at the time a patient is accepted for admission.
The patient is eligible for randomisation (see above).
3.The ICU floor coordinator or responsible clinician on duty opens the next envelope in sequence to reveal allocation of the patient to an intervention or control bedspace.
4.The patient is assigned to the allocated bed.
5.Patients will then be assessed for eligibility for participation in the study by a member of the research team (see ‘Recruitment’ section below).
Case–control sub-studyBedspaces in the study ICU differ regarding access to windows and natural light, potentially confounding outcomes. To investigate the effect of these variables, this study involves a concurrent observational case–control study utilising the three bedspaces that are most similar to the intervention bedspaces. These three internal and windowless bedspaces will function as ‘environmental control’ bedspaces. Thus, all patients admitted to these three bedspaces will also be invited to participate using the same protocols as the main RCT, but analysis will be adjusted to account for any potential bias in admission diagnosis, severity of illness, or other baseline data compared to the RCT cohort. Where patients are allocated to the environmental control bedspaces as part of the RCT, no further action will be taken, but patient data will also be utilised for the independent sub-analysis comparing intervention to environmental control bedspaces. Additionally, patients admitted to the intervention beds without being randomised will be approached for participation in the study as case control patients.
RecruitmentFor an estimated study period of 2 years, all patients randomised to an intervention or control ICU bedspace as part of the RCT or admitted to environmental control bedspaces as part of the case–control study will be screened for suitability to participate in the research study. Suitable patients will be approached by a member of the research team as soon as possible after admission to ICU, in consultation with the treating clinical team. As patient status commonly changes throughout ICU admission, patients may be screened daily until discharge and may be recruited to the study at any time during their admission to ICU if suitable based on the following inclusion/exclusion criteria:
Inclusion criteriaPatient residing in Australia
Patients sufficiently fluent in English to complete recruitment and data collection processes
Patients expected to remain in ICU for > 24 h
Exclusion criteriaPatient or legal representative unable or unwilling to provide consent.
Patients less than 18 years of age.
Death is deemed imminent.
Patient deeply or moderately sedated (RASS score ≤ − 3).
Recent substantial neurological insult (e.g. stroke).
Patient deemed agitated, aggressive, or displays unpredictable behaviour.
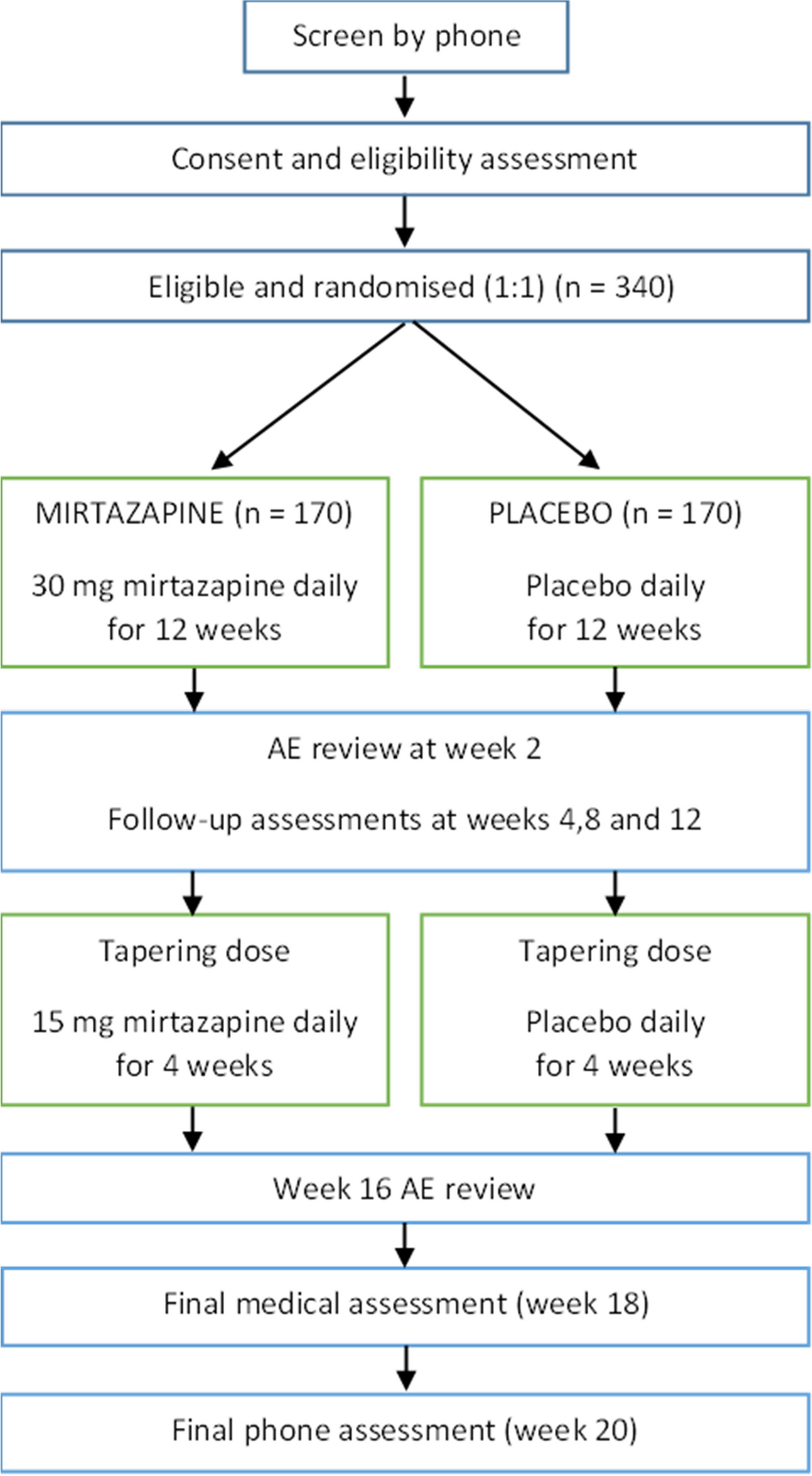
Patients will be informed of the purpose and aims of the study and informed consent will be obtained prior to being enrolled in the study. If patients are unable to consent, the substitute decision-maker or next-of-kin will be approached to consider participation. Figure 2 summarises the enrolment, intervention, and assessment timeline.
Fig. 2
Study schedule of enrolment, intervention, and assessments
Patients for whom consent is obtained will complete the study components listed below. Based on annual admission numbers and current admission diagnoses and presentations, the average number of patients eligible for randomisation per bedspace in the study facility is approximately 30 per year. We therefore estimate that approximately 250 patients will be randomised over 2 years. We expect that approximately 50 patients will be ineligible for participation for various reasons and based on other studies in the unit we have conservatively estimated a 50% patient decline rate. We therefore estimate that approximately 100 patients will provide consent and be recruited to this study over the 2-year study period.
A power calculation for this study is not possible, due to the lack of studies reporting the impact of environmental upgrades on patient outcomes. Therefore, an interim analysis will be conducted after 6 months of data collection, focussing on recruitment rate, completion rate for the various study components, and in-hospital outcome measures, with a power calculation performed based on this data to determine the number of participants required.
Data collection and monitoring, study components, and data analysisData describing participant demographics and ICU admission will be collected from hospital records and recorded on case report forms (CRFs). Data collected will include date of birth, gender, hospital and ICU length of stay, ICU and hospital survival, ICU readmission rates, mechanical ventilation time, details of their illness, medical treatment, and existing comorbidities. Completed CRFs will be entered into an electronic database. Patient characteristics and ICU admission data will be described using summary statistics. The number of patients approached and consented will be charted, together with the number who dropped out or died.
A data monitoring committee was not considered as this was a low-risk intervention. Data monitoring and study conduct is reviewed 6 monthly by the research investigators and submitted for ethics committee review yearly.
In addition to data from the study components being individually analysed and reported as described below, data analysis will also investigate the relationships between different outcomes/components. Inter-related components that will be explored include whether observed differences in sleep architecture is correlated with changes in delirium outcomes, and whether a difference in the maintenance of circadian rhythm between intervention and control bedspaces impacts the incidence of PICS at 6-month post-ICU discharge.
Component 1: objective environmental assessment of the upgraded ICU environmentThis observational component of the study has been designed to generate a comprehensive quantitative account of the upgraded ICU environment with reference to physical space, sound, acoustics, lighting, temperature, and humidity, and compare with previously collected baseline/control data. The intervention bedspaces will be continuously monitored for the study period, with sound levels (dB/dBA), light levels (lux), temperature (°C), and humidity (%) all being monitored via wireless integrated environmental monitoring sensors (Senseagent LP4-204 Hilbert Sensor). These sensors communicate via a wireless LP4-391 Neumann Gateway, with members of the research team able to download the data from a cloud-based database into an Excel spreadsheet. The evaluation of sound levels will also include audits of the number and type of alarms, with alarm data downloaded from key equipment in ICU, including patient monitors and ventilators.
A detailed acoustic and lighting evaluation will be performed. A specialist acoustic technician will perform digital acoustic testing of the intervention ICU bedspace environment. Photometric measures of the lighting environment will be collected over a 48-h period. Measurements will include indoor vertical and horizontal illuminance (photopic and melanopic), luminance, and reflectance values at both room and patient level. Measures will be collected with unobtrusive, validated, and calibrated instruments, including wearable devices to evaluate the lighting quality and quantity as experienced by patients.
Continuous data, such as sound levels, light levels, temperature, and humidity, will be described using mean and standard deviation and compared to previously collected bedspace data using a one-sample t-test. Daytime and night-time data will be evaluated and compared. The number of alarms will be described using their frequency and compared to previously collected data using chi-squared tests.
Component 2: stakeholder perspectives: patients, relatives, and staffThis component of the study will use qualitative methods to describe how the redesigned ICU bedspaces influence the patient experience in ICU from the perspective of patients, family members, and staff. Patients admitted to the intervention bedspaces who survive their ICU admission will be invited to take part. Data will be collected in semi-structured interviews with individual patients, accompanied by family members when available and willing to take part. Interviews will be conducted within a week after discharge from ICU but before hospital discharge. Staff from medical, nursing, allied health disciplines, and non-clinical/operational staff providing a range of services in the ICU will be invited to participate in focus groups or individual interviews. The interviewer will use a topic guide flexibly to explore participants’ experiences in the intervention bedspaces. Based on our previously conducted baseline studies [4, 31], we anticipate that between 10–15 patients and 25–30 staff will be required to reach saturation and address research questions.
The approach to analysis of the qualitative interviews has been described in detail elsewhere [4, 31]. In brief, recorded interviews will be transcribed and analysed using a framework approach [35]. Framework analysis consists of five steps: (1) familiarisation, (2) identifying thematic framework, (3) indexing, (4) charting, and (5) mapping and interpretation.
Component 3: sleep quality and quantityThe quality and quantity of participants’ sleep will be measured objectively, using a combination of single forehead sensor electroencephalogram (EEG) and polysomnography, as well as subjectively using validated sleep questionnaires. Sleep studies will only be commenced when the effects of relevant medications are no longer likely to impact on the quality of data collected; therefore, sleep will not be measured on participants that are deeply or moderately sedated (RASS score − 3, − 4, or − 5), or have had a general anaesthetic, drug overdose, or alcohol intoxication in the preceding 24 h. Ventilated and non-ventilated participants will be included.
Sleep will be measured continuously for a 2–4-day study period (dependent on duration of ICU admission) following recruitment using the Somfit single forehead sensor EEG (Compumedics®). This is a recently developed wearable device that is light and comfortable for patients to wear while enabling collection of high-quality EEG signals.
Sleep will be concurrently measured using a gold-standard portable polysomnography (PSG) recorder for a 24-h period. Polysomnography involves the application of sensors and electrodes for the continuous monitoring of physiological variables during sleep. The portable PSG is a small device that is worn as a belt across the patient’s thorax. Ten sensors are attached to the patient: 2 under the chin, 1 next to each eye, and 6 on the scalp. The sensors will measure and monitor muscle tone changes using electromyogram (EMG), eye movements using electrooculogram, and electrical activity in the brain using EEG. The equipment also has an inbuilt oximeter to measure pulse oximetry and can also measure patient body position as well as background light and sound. The equipment also collects electrocardiogram, leg EMG, nasal pressure/thermistor, and ribcage/abdominal movements.
Participants will be asked to complete the validated sleep in the ICU questionnaire and the Morningness-Eveningness Questionnaire before discharge from ICU [36, 37]. The aim of these questionnaires is to establish the patients’ reported quality and quantity of sleep as well as the reasons for sleep disruptions.
The data will be analysed and interpreted by expert sleep scientists. Objective measures of sleep will be described for a minimum 24-h period, including total sleep time, time awake, sleep staging (stage 1 and 2, slow wave sleep, and rapid eye movement (REM) sleep), arousal index, number of awakenings across the 24-h period, and percentage of sleep at night-time versus daytime hours. Objective sleep quality will be compared between control and interventional beds.
Component 4: circadian rhythmsFor this component, data will be collected from environmental sensors, study records, participants, medical records, and biological samples. The following measured variables will be analysed to evaluate the circadian synchronisation of the patients and study their influence on the outcome of the patients:
Routinely collected physiological data, including body temperature, heart rate and heart rate variability, blood pressure, pulse oximetry, and enteral/parenteral feeding rhythm
Relevant medications with the potential to modify patients’ heart rate and blood pressure
Quality and quantity of sleep (as described for component 3 above)
Four-hourly blood samples for 48 h, specifically looking at cortisol, melatonin, insulin-like growth factor 1, inflammatory markers (full blood count and c-reactive protein), haemoglobin, proteomics analysis, and expression of circadian clock genes and untargeted RNA-sequencing in white blood cells
Biological measured variables will be assessed using a modified version of dryR, a statistical framework to assess differential rhythmicity [38]. The analysis is based on multiple mixed linear regression with a subsequent model selection approach based on the Bayesian information criterion to assess differential rhythmicity of the measured variables, comparing patients admitted to the intervention versus control beds. The mixed linear models will include fixed effects from a harmonic regression model and a random effect (patient–specific) on the intercept that deals with the subject–to–subject variation and dependency of the repeated measures. Rhythmic parameters including amplitude and acrophase will be computed from the selected model for each of the conditions. This will identify circadian/diurnal variables that are impacted by the intervention.
Component 5: delirium prevalenceThe prevalence of delirium will be assessed by the completion of the confusion assessment method for the intensive care unit (CAM-ICU) twice per day (morning and afternoon) for all enrolled patients by a member of the research team.
Delirium data analysis will involve:
1.The prevalence and incidence of delirium will be calculated and compared between intervention and control bedspaces.
2.Comparison of cases and non-cases on demographic and diagnostic data and length of stay will be undertaken using methods appropriate to type of data: T-tests, chi square, and ANOVA.
Component 6: medium-term outcomes after ICU admissionData on patient outcomes after ICU discharge will be collected from participants via a battery of validated self-report tools 6 months after ICU discharge. The method used for this is summarised in Table 2.
Table 2 Questionnaire summaryFor this study, a case of PICS will be defined as any participant with questionnaire scores outside of normal ranges indicating impaired physical (EQ-5D-5L), psychological (HADS or PCL-5), and/or cognitive function (PROMIS). The incidence of PICS will be described using proportions and associated 95% confidence intervals and compared between sub-populations, such as ICU stay < 48 h, planned vs. emergent admission, ventilated vs. non-ventilated, and type of bedspace admitted to. Multivariable regression analysis will be used to identify factors associated with PICS, or not getting PICS. The overall characteristics of the sample will be described using summary statistics. This will help inform the generalisability of our results. The number of patients approached and consented will be tabulated, together with the number who were lost to follow-up, withdrew from the study, or died.
Component 7: health economic evaluationA within-trial cost-effectiveness analysis will be conducted to estimate the changes to total costs and patient outcomes associated with the intervention. The change to total costs will reflect the cost of implementing the intervention under different scenarios, offset by the economic value of ICU bed days saved from reduced ICU length of stay [43]. Planned scenarios will consider different definitions of implementation costs per intervention bedspace, which will impact the change in total costs per unit of health benefit. Definitions will reflect the cost of maintaining a bedspace that already exists in the ICU (base case), the cost of adding a new bedspace within an existing ICU, and the cost of building a new ICU to accommodate the intervention. These costs will be measured from the health system perspective. We will further consider the economic value of lost productivity from the patient perspective, which will be collected using the patient employment information questionnaire. Patient outcomes will be measured in quality-adjusted life years, which reflect patient life expectancy adjusted for health-related quality of life. Life years will be estimated using Australian life tables based on patients’ sex and age at the time of ICU admission. Quality of life, measured under component 6 via the EQ-5D-5L, will be used to estimate health utility values needed to convert life years to quality-adjusted life years.
Cost-effectiveness outcomes will be reported as an incremental cost-effectiveness ratio and net monetary benefit, for different willingness to pay thresholds. The impact of model input uncertainty on cost-effectiveness outcomes will be examined by deterministic sensitivity analysis and probabilistic sensitivity analysis. Model input distributions needed to conduct sensitivity analyses will be informed by available study data, published literature, and expert opinion where appropriate. Full details of analysis methods, assumptions, and results will be transparently reported in accordance with the 2022 Consolidated Health Economic Evaluation Reporting Standards (CHEERS 2022) checklist.
留言 (0)