
記住我



In humans, growth factor receptors (GFRs) superfamily includes proteins capable of transduce extracellular signals to cellular responses and in particular to prompt mitogenic activity of growth factor ligands. Heterogeneous molecular signals are conveyed across the cytoplasm to the transcriptional machinery into the nucleus through sequential kinase signaling, settled in redundant and cross-talking cascades [1, 2]. GFRs are sited in the cell plasma membrane as monomers or (pre)dimers and ligand binding elicits higher-order oligomerization of ligand-receptor complexes. GFRs encompass three domains: (i) extracellular ligand (growth factor) binding domain, (ii) transmembrane domain, (iii) cytoplasmic domain with enzymatic activity or complexing with enzymatic proteins [1, 2]. The expression of genes encoding GFRs is positively or negatively regulated at the transcriptional level through protein-DNA and protein–protein interactions in various cell types, depending on the developmental and cellular context. Establishment of the ligand-receptor complex through interaction of the three-dimensional structure of ligands with the related members of the GFR tyrosine kinase (RTK) or serine-threonine kinase (RSTK) superfamily initiates the signal transduction cascade via auto-phosphorylation by the intracellular tyrosine or serine-threonine kinase domain [1, 2]. The organization of the extracellular portion of the receptor enclosing the ligand binding domain differs considerably among the different superfamily members and binding of growth factors to their cell plasma membrane receptors elicits phosphorylation of tyrosine or serine-threonine residues on numerous intracellular signaling molecules, which transmit the signal in the cytoplasm and nucleus [1, 2]. Mainly, GFRs signaling through RTKs actuate molecular cascades such as phosphatidylinositol 3-kinase (PI3K) pathway, mitogen-activated protein kinase (MAPK) via the Rat sarcoma/rapidly accelerated fibrosarcoma/mitogen-activated protein kinase (RAF-MKK1/2-Erk1/2/MAPK kinase cascade) and JAK2/STAT signaling. RSTKs encompass a set of related catalytic receptors comprising TGF-β receptors (TGF-βRI-III) and activin receptors (ALK1-7) existing as heterodimers of type I and type II receptors, with the ligand binding domain located in the type II receptor [1, 2]. Upon ligand-receptor binding, the type I receptors are recruited to the complex and are phosphorylated by the type II receptor [1, 2]. Activated type I receptors subsequently phosphorylate SMAD proteins, depending in detail on the RSTK subtype: TGF-βR and ALK4/5/7 phosphorylate SMAD2/3, whereas ALK1/2/3/6 phosphorylate SMAD1/5/8. Upon activation, SMAD proteins complex with SMAD4 and after conveyance into the nucleus bind transcription factors and co-factors to control expression of target genes [1, 2]. A wide-ranging pathway atlas for EGFR-mediated signaling comprises EGFR endocytosis with sequential degradation or recycling, small guanosine triphosphatase (GTPase)-mediated signal transduction, MAPK and PI3K signaling, and G protein-coupled receptor (GPCR)-mediated EGFR transactivation through intracellular Ca2+ signaling [1, 2]. Deranged intracellular signaling plays a crucial role in oncogenesis and is a target of therapeutic interventions [3,4,5,6]. The expression of genes encoding GFRs and their specific ligands oscillates rhythmically and show patterns defined circadian (from the Latin words circa and dies, approximately 24 h) [7]. Circadian rhythms of biological processes are driven by the circadian clock circuitry, a hierarchical organization of molecular clockworks ticking in every cell of the body and working through a molecularly hardwired negative feedback loop with temporal delay [8].
The circadian clock circuitryOn our planet, life forms adjusted to 24-hour light/darkness and temperature transition and seasonal rhythmicity, related to Earth’s spin on its axis and revolution motion around the Sun, respectively, developing endogenous molecular clockworks to synchronize their behavioral and metabolic rhythms to predictable environmental cues [9, 10]. The harmonized regulation of sleep/wake, rest/activity, fasting/feeding cycles with biochemical processes and metabolic pathways is crucial to maintain body homeostasis and preserve health. Coordinating biological rhythms and anticipated changes of spatio/temporal niches impacts organism and species survival of free-living animals, influencing behavioral cycles of feeding, predation, competition, mating and providing fitness advantage as opposed to natural selection pressure [11,12,13]. Loss of resonance of body rhythmicity with external synchronizers or a change of oscillation phase may alter the physiological array of rhythms with onset of internal desynchronization, also known as chronodisruption, which promotes neoplastic, metabolic, inflammatory and neurodegenerative diseases [11,12,13]. Daily timekeeping is driven in mammals and humans by the circadian timing system, comprising a central pace-maker and master oscillators located in the suprachiasmatic nuclei (SCN) of the hypothalamus and autonomous self-sustained oscillators in every cell of peripheral tissues. Ambient natural or artificial photic cues are perceived by non-image forming intrinsically photosensitive retinal ganglion cells containing melanopsin, whose output is transferred to the SCN by the retino-hypothalamic tract [14,15,16]. SCN coordinate brain nuclei and peripheral oscillators through neural and hormonal signals, represented by autonomic nervous system innervation and circulating systemic factors (cortisol during the day and melatonin at night) [17]. The biological oscillators in every cell are driven by molecular clockworks ticking through a set of genes whose expression oscillates with circadian rhythmicity, as well as the encoded proteins that in turn block their transcriptional activation, operating a loop in which the product of gene transcription in sequence and with delay decreases gene expression [14,15,16]. Peripheral oscillators are entrained also by food assumption and fluctuation of nutrient levels, while central oscillators in the SCN are resilient to feeding-related stimuli and are entrained primarily by light/darkness alternation, so that desynchronization of food intake and metabolic processes respect to proper diurnal activity patterns uncouple the light-driven SCN and tissue/organ systems oscillators [14,15,16].
The molecular clockworkThe molecular cogs operating to drive circadian rhythmicity at the cellular level are represented by a group of genes, named core clock genes, and their encoded.
proteins that manage interlocking transcription-translation feedback loops (TTFLs), in addition to non-transcriptional loops, carrying out one cycle in roughly 24 h [18,19,20,21]. The TTFL is worked by a positive limb, operated by the bHLH-PAS (basic helix-loop-helix–Period-Arnt-Single-minded) transcriptional activators CLOCK (circadian locomotor output cycles kaput), and its paralog NPAS2 (neuronal PAS domain protein 2), and BMAL1-2/ARNTL-2 (brain and muscle aryl-hydrocarbon receptor nuclear translocator-like/aryl-hydrocarbon receptor nuclear translocator-like) that heterodimerize and bind to enhancer (E)-box DNA consensus sequences of the target Period (PER1-3) and Cryptochrome (CRY 1–2) genes [18,19,20,21]. The encoded PER1-3 and CRY1-2 proteins operate the TTFL negative limb; they accrue and dimerize in the cytoplasm forming repressor complexes that pass back into the nucleus and inhibit the transcriptional activity of CLOCK: BMAL1-2 heterodimers [18,19,20,21]. The circadian proteins undergo post-translational modifications (PTMs), such as phosphorylation, acetylation, sumoylation, O-GlcNAcylation) modulating their activity and in sequence ubiquitination/proteasomal degradation allowing correct functioning of the TTFL and setting of biological clock speed [22,23,24]. An auxiliary interconnected loop is operated by the nuclear receptors (NRs) REV-ERBs and retinoic acid-related (RAR) orphan receptor (RORs), which drive BMAL1 rhythmic transcription competing for a ROR specific response elements (RORE) in its promoter [25]. ROR-α works as transcription activator and physically interacts with peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α (PGC-1α), which recruits chromatin-remodelling complexes to proximal BMAL1 promoters and elicits BMAL1 transcription. Conversely, REV-ERB-α interacts with the nuclear corepressor/histone deacetylase3 (NCoR-HDAC3) corepressor complex and inhibits BMAL1 transcription [25]. Other than the NR operated feedback loop, CLOCK: BMAL1 heterodimers through cognate D-box elements regulate the expression of the PAR domain basic leucine zipper (bZIP) transcription factors and first order clock controlled gens DBP (albumin D-site binding protein), TEF (thyrotroph embryonic factor), HLF (hepatic leukaemia factor), which successively drive the rhythmic expression of thousands tissue specific (output) genes [26, 27]. Besides, REV-ERBs and DBP competing for Res drive the expression of the Nuclear factor, interleukin 3 regulated protein (NFIL3, also known as E4BP4), whose promoter contains a RORE, so that its transcription is suppressed by REV-ERBs and shows an oscillatory pattern with opposite phase respect to DBP [26, 27]. Other circadian components involved in the molecular clockwork are the E-box-binding basic helix-loop-helix transcription factors DEC1 (Differentially expressed in chondrocytes protein 1) and DEC2 [26, 27]. In particular, DEC1 transcription is elicited by CLOCK: BMAL1 heterodimer, but in turn DEC1 proteins repress CLOCK: BMAL1 transcriptional activity, operating a power steering feed-back [26, 27]. Epigenetic modifications fine tune the functioning of the molecular clockwork through rhythmic chromatin-histone remodelling and are mainly primed by acetylation/deacetylation as well as methylation/demethylation processes. Regarding the cogs of the molecular clockwork, BMAL1 acetylation is operated by CLOCK, which has intrinsic protein and histone acetyltransferase capability [28]. SIRT1, a type III histone/protein deacetylase, counters this process and its activity depends on the intracellular levels of nicotinamide adenine dinucleotide (NAD+), a nutrient sensor synthesized from tryptophan through the enzymatic activity of nicotinamide phosphoribosyl-transferase (NAMPT/visfatin), whose expression is rhythmically driven by the biological clock [29,30,31,32]. (Fig. 1).
Fig. 1
Outline of the molecular clockwork. The biological clock is hard-wired by interlocking transcriptional–translational feedback loops (TTFLs) operated by a set of circadian protein-encoding genes. The positive limb of the TTFL is ran by the transcription factors BMAL1 or its analog BMAL2 (schematically rendered as BMAL) and CLOCK, which heterodimerize and bind to E-box enhancer elements in the promoters of the Period genes (PER1-2) and Cryptochrome genes (CRY1-2). PER and CRY proteins operate the negative limb of the TTFL and hinder the transcriptional activity of BMAL:CLOCK heterodimers. The nuclear receptors REV-ERBs and RORs compete at specific response elements (RRE) on the promoter of BMAL1 and drive its rhythmic expression. See text for in depth explanation and details
Epidermal growth factor receptor-dependent signalingIn mammalian cells the Epidermal Growth Factor Receptor (EGFR) signaling pathway controls essential functions such as survival, proliferation and migration. Different subtypes of erythroblastic leukemia viral (v-erb-b) oncogene homolog (ErbB) receptors bind a family of ligands with activation of various molecules in a wide-ranging system of receptor complexes expressed on near every human cell type [33]. Ligand binding prompts homo- and hetero-dimerization of four ErbB family receptors: ErbB1 (also known as EGFR), ErbB2, ErbB3, and ErbB4 [2]. EGFR ligands comprise several molecules, among which transforming growth factor (TGF)-α, amphiregulin, heparin-binding EGF-like growth factor (HB-EGF) and epiregulin, decisively entailed in tissue-specific proliferation/differentiation homeostasis. Binding of ErbB dimer by a definite ligand elicits ErbB cytoplasmic kinase activity and starts auto- and transphosphorylation on tyrosine residues, providing a docking site for adaptor proteins and enzymes, while the ATP binding pocket is held between the two lobes of the kinase fold [2]. EGFR activation may occur with numerous mechanisms under physiological or pathological conditions. In addition to direct activation by specific ligands, heterologous ligand-dependent mechanisms are likewise involved, such as EGFR activation through G-protein-coupled receptors (GPCRs) activation by mature forms of EGFR ligands cleaved by metalloproteinase from membrane precursors [2].
EGFR ligand-independent mechanisms may be also involved. For instance, integrins may form physical complexes with GFRs at the cell plasma membrane. Also, transactivation from GPCR cytokine receptors may play a role, regulating pro-inflammatory activation and transducing modifications in proliferation rate as well as cellular shape and attachment [2].
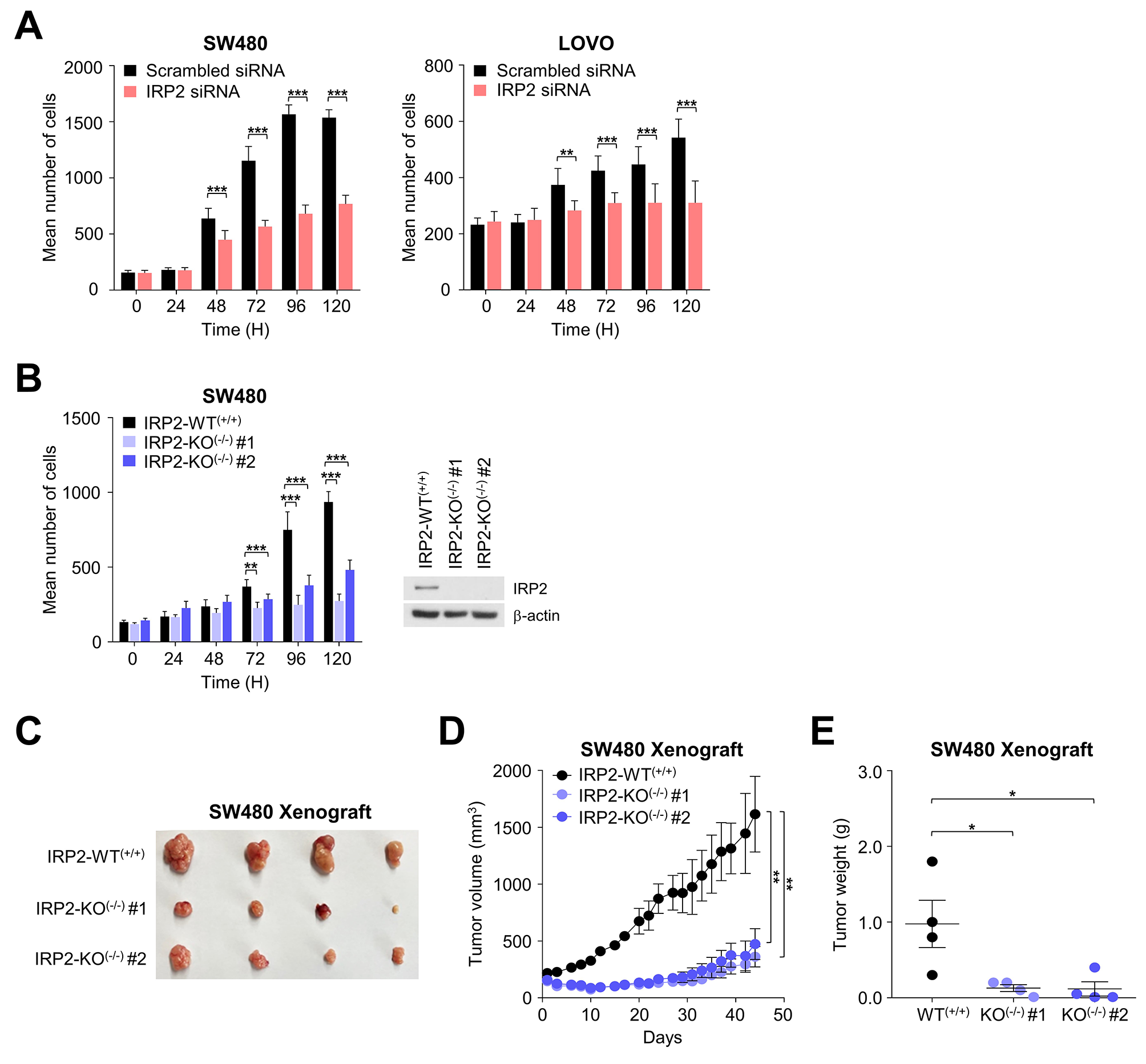
Conversely, EGFR functions can be constrained through de-phosphorylation by active protein-tyrosine phosphatases, which sequentially may be reversibly inactivated through oxidation of the catalytic cysteine residue in their active site by reactive oxygen species produced upon EGFR activation [2]. The EGF family mediators bind with autocrine/paracrine pattern their receptors through the ligand binding site on the extracellular domain of cell plasma membrane receptors and elicit an active reparative response in case of biophysical damage, but can also play a role in deranged cell processes involved in carcinogenesis. Ligand binding prompts homo- and hetero-dimerization of the receptor inducing intracellular RTK activation. On their side, the IGF-1 and insulin receptors exist as covalent cross-linked dimers with each monomer comprising two subunits [2]. Growth factor signaling can be terminated by receptor-mediated endocytosis with internalization of the ligand-receptor complex, but in case of oncogenic changes RTKs signal unrelenting cell proliferation regardless of growth factor stimulation [34]. In human cancers constitutive RTK activation is initiated by gain-of-function mutations, genomic amplification, chromosomal rearrangements, and autocrine activation. Mutated forms of growth factor proteins with derangement of GFR activation hallmark virtually all epithelial cancers, but on the other hand represent valuable targets for therapeutic agents [34].(Fig. 2; Table 1).
Fig. 2
Schematic representation of intracellular signaling prompted by Receptor Tyrosine Kinases (RTKs) and Receptor Serine/Threonine Kinases (RSTKs). See text for in depth explanation and details
Table 1 List of receptor tyrosine kinasesGFR-dependent signaling and circadian pathways in the context of carcinogenesisCancer onset and progression is fueled by complex molecular mechanisms involving genetic and epigenetic changes at the level of driver oncogenes and/or controller tumor suppressor genes leading to deregulated activation of key signaling pathways, many of which are controlled by the biological clock and show circadian patterns of activation, for instance PI3K/AKT/mTOR, MAPK and JAK/STAT signaling pathways, crucially involved in the regulation of cell growth/survival, proliferation and differentiation.
PI3K/AKT and mTOR signaling pathways and the biological clockThe PI3K/AKT pathway operates downstream of EGFR and human epidermal growth factor receptor (HER)2 controlling essential cell processes comprising growth, proliferation, survival, and motility/migration. Deranged PI3K/AKT signaling pathway activation upkeeps cancer cell proliferation and survival, favoring metastatic spread and chemoresistance [35]. On its side, AKT serine/threonine kinase comprises various domains: (i) N-terminal pleckstrin homology (PH) domain, a sequence of 100 amino acids interacting with phosphoinositides produced by the lipid kinase phosphoinositide 3-kinase (PI3K), AKT (ii) kinase domain, located at the heart of the molecule and similar to other Ser/Thr protein kinases of the AGC kinase group, counting the cyclic-nucleotide-dependent family (PKA and PKG), the protein kinase C family, the β-adrenergic receptor kinase (βARK), the ribosomal S6 family among others; (iii) C-terminal regulatory domain, a sequence of approximately 40 amino acids including the hydrophobic motif, whose phosphorylation initiates enzymatic activity [35]. Upon activation, AKT limits cell apoptosis and growth promoted by TGF-β and C/EBPα, respectively, and may turn on β-catenin downstream signaling. PI3K/AKT and TGF-β/SMAD signaling pathways interplay to control several cell processes, counting proliferation, apoptosis, and migration: TGF-β/SMAD signaling exerts cytostatic activity in premalignant states opposing pro-cell survival effect exerted by growth-factor-mediated PI3K/AKT activation, so that the two pathways might oppose each other in balancing cell survival [36]. In advanced cancers the anti-proliferative action of TGF-β is jammed and both TGF-β receptor-dependent and PI3K/AKT pathways exert pro-oncogenic effects through complex signal integration and obligated cooperation to support cancer development and progression [36]. The biological clock is synchronized by external and internal stimuli to cope with modifications in the environment or in the internal milieu and PI3K-dependent intracellular-signaling pathway was shown able to play a role in the fine-tuning of the molecular clockwork targeting BMAL1 and CLOCK and altering circadian DBP gene expression. PI3K knockdown by pharmacological inhibitors or shRNA-mediated silencing hindered DBP mRNA upregulation in NIH-3T3 cells (fibroblast cell line isolated from NIH/Swiss mouse embryo) synchronized with serum shock (treatment of cultured cells with high concentrations of serum) [37]. Furthermore, PI3K inhibition blocked BMAL1 and CLOCK heterodimerization, considerably decreased DBP gene promoter activity and diminished BMAL1/CLOCK heterodimer recruitment to the E-box DNA response element in the DBP promoter, with consequent transcriptional modulation of this important first order clock controlled gene [37]. The interplay of PI3K and circadian pathways was evaluated in the neural framework of cellular survival and a modulatory effect of BMAL1 protein together with PI3K/AKT signaling cascade regulation was shown, in addition to a supporting effect of melatonin on BMAL1 expression. BMAL1 and melatonin increased cellular survival after oxygen glucose deprivation with concurrent phosphorylation of AKT, ERK-1/2, PDK1, mTOR, PTEN, GSK-3αβ, and p70S6K, while PI3K/AKT inhibition diminished BMAL1 expression, suggesting that melatonin modulates BMAL1 expression through PI3K/AKT signaling and BMAL1 hampers cell death triggering survival kinases [38]. The PI3K/AKT pathway cooperatively interplays with the serine-threonine kinase mammalian target of rapamycin (mTOR), importantly involved in mRNA translation and protein synthesis [39, 40]. Remarkably, the circadian clock circuitry and mTOR signaling pathway interact at many levels and mTOR cascade rhythmic activation is driven by the biological clock in the SCN in mice [41]. Monitoring of the mTOR activity marker phosphorylated S6 ribosomal protein (pS6) evidenced maximal activity of the mTOR pathway during the subjective day and minimal activity during the late subjective night [41]. The circadian protein Per2 limits mTORC1 complex activity binding specifically and conjointly tuberous sclerosis complex 1/2 (TSC1/2), Raptor, and mTOR. On the other hand, PER2 expression is set off by the glucagon-Creb/Crtc2 signaling, with consequent mTORC1 suppression and
留言 (0)