Data collection and processing
The acquisition and processing of RNA-seq data and clinical data from patients with Liver Hepatocellular Carcinoma (LIHC) were conducted using resources such as The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov), Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/gds), and International Cancer Genome Consortium (ICGC, https://dcc.icgc.org/). The expression data for liver cancer cell lines were sourced from the DepMap portal (DepMap, https://depmap.org/portal/). Differential expression analysis was performed on LIHC samples, distinguishing between tumour and normal samples. Among the 33 types of tumours, only 17 types contained more than five pairs of tumour and normal samples. Consequently, differential expression analysis was restricted to these 17 types, with the ratio of the average expression value of tumour samples to that of normal samples representing the fold change. The t-test was employed to derive the associated p-value.
Upon integrating the expression data of tetraspanin-related genes with clinical information, risk ratios relative to Overall Survival (OS) were calculated for LIHC using Cox regression analyses to categorise samples into high or low risk. LIHC samples were dichotomised into two groups based on the optimal cutoff value, and a log-rank test was utilised to determine the associated p-value. Kaplan-Meier survival curves for OS were constructed for tetraspanin-related genes significant to LIHC, and subjected to the log-rank test. The graphical representation was achieved using the R package “survival.”
Cell culture and transfection
HCCLM3 and MHCC97H cells were established at the Liver Cancer Institute (Zhongshan Hospital, Fudan University, Shanghai, China), and Hep3B, Huh7, HepG2 cells were procured from the American Type Culture Collection (ATCC). These cell lines, with STR identification reports, were consistently maintained.
To establish the HCCLM3-TSPAN4-GFP stable cell line, HCCLM3 cells underwent transfection with lentivirus, followed by selection with Blasticidin. The HCCLM3-CD151-RFP, HCCLM3-TSPAN4-RFP, Hep3B-TSPAN4-RFP stable cell lines were generated by transfecting HCCLM3 cells with CD151-RFP/TSPAN4-RFP vector, and Hep3B cells with TSPAN4-RFP vector, followed by selection with puromycin.
Animal experiments
Six-week-old male athymic BALB/C nu/nu mice were randomly allocated to groups prior to inoculation. Mice were housed in a standard animal laboratory with unrestricted access to food and water, under constant environmental conditions with a 12-hour dark-light cycle. CD151 shRNA or Mock shRNA infected HCCLM3-luc/MHCC97H-luc cells (1 × 10^7) suspended in 0.2 mL serum-free culture medium were injected into the upper flank region of nude mice. Sacrifice occurred four weeks later, and tumors were harvested and measured for volume. Tumor volume was calculated using the formula: volume (mm^3) = (width)^2 × length/2.
An orthotopic model was constructed by producing tumors as described above. After two weeks, tumors were sectioned into small pieces of approximately 1.0 mm^3 and orthotopically transplanted into the livers of nude mice. These mice were allowed to grow for three months, and D-Luciferin was intraperitoneally injected. The emitted photon signal, detectable using Bruker MS FX PRO In vivo Imager, facilitated quantification of the total tumor burden. This technology enabled the assessment of tumor development ex vivo. The colour gradient represented the in vivo size of the tumor. Mice were then euthanised by cervical dislocation, and the livers were resected and photographed using a high-definition digital camera. All procedures complied with the Animal Committee of Tongren Hospital guidelines (approval number: A2023-119-01). The animal experiments in this study were strictly conducted in accordance with the guidelines provided by the Ministry of Science and Technology of the People’s Republic of China regarding the humane treatment of experimental animals, among other regulations.
Preparation of small interfering RNA (siRNA)
CD151 (Human) siRNA were synthesized by KeyGEN BioTECH (China). Sequences of the three synthesized oligonucleotides are: CD151si1 sense: 5′-GCUGGAGAUCAUCGCUGGUAUTT-3′, CD151 si1 antisense: 5′-AUACCAGCGAUGAUCUCCAGCTT-3′; CD151si2 sense: 5′- CCCUCAAGAGUGACUACAUCATT-3′, CD151 si2 antisense: 5′- UGAUGUAGUCACUCUUGAGGGTT-3′; CD151si3 sense: 5′- GCCUCAAGUACCUGCUGUUUATT-3′, CD151 si3 antisense: 5′- UAAACAGCAGGUACUUGAGGCTT-3′.
RNA interference, RNA isolation, and real-time PCR
Plasmids expressing short hairpin RNA (sh-RNA) targeted the following sequences: CD151-shRNA-1: 5′-GCCTCAAGTACCTGCTGTTTA-3′; CD151-shRNA-2: 5′-ACCTGCTGTTTACCTACAATT-3′; CD151-shRNA-3: 5′-CCCTCAAGAGTGACTACATCA-3′. A non-target shRNA (Scramble shRNA) against the human genome served as the control. Total RNA was extracted from cells using Trizol reagents (Invitrogen, Shanghai). The isolated mRNAs were reverse transcribed into complementary DNA (cDNA) using the Promega Reverse Transcription System (Madison, WI, USA) with Oligo dT priming. Real-time PCR was conducted using SYBR Green Premix Ex Taq (Takara, Japan) on a Light Cycler 480 (Roche, Switzerland). GAPDH mRNA levels were utilised as an internal control. Gene expression differences were determined using the 2-ΔΔCt method and expressed as fold-changes. PCR conditions comprised an initial holding period at 95 °C for 5 min, followed by a two-step PCR program of 95 °C for 5 s and 60 °C for 30 s for 50 cycles. Q-PCR primer sequences for CD151 were as follows: forward, 5′- AGACCATGCCTCCAACATCTA-3′, and reverse, 5′- CACAGGCAATGCCGATCC-3′. GAPDH primer sequences for Q-PCR were as follows: forward, 5′- AGGTCGGAGTCAACGGATTTG′, and reverse, 5′- CATCGCCCCACTTGATTTTG-3′.
Western blot
Total proteins were extracted using the Whole Protein Extraction Kit (KGP200, KeyGen BioTECH, China), and protein concentrations were determined using a BCA protein assay kit (KGPBCA, KeyGen BioTECH, China). Separated samples were subjected to 10% SDS-PAGE and subsequently transferred to PVDF membranes. The following antibodies were used: CD151 (1:1000, sc-271,216, Santa Cruz Biotechnology, USA), β-actin (1:1000, sc-47,778, Santa Cruz Biotechnology, USA), EOGT (1:1000, ab190693, Abcam, USA), TSPAN4 (1:1000, ab181995, Abcam, USA), TSPAN7 (1:1000, 18695-1-AP, Proteintech, USA), Integrin α5 (1:1000, 4705, Cell Signaling Technology, USA), PIGK (1:1000, ab201693, Abcam, USA), PGCP (CPQ) (1:1000, ab96159, Abcam, USA), CD9 (1:1000, sc-13,118, Santa Cruz Biotechnology, USA), CD63 (1:1000, sc-5275, Santa Cruz Biotechnology, USA).
Confocal imaging
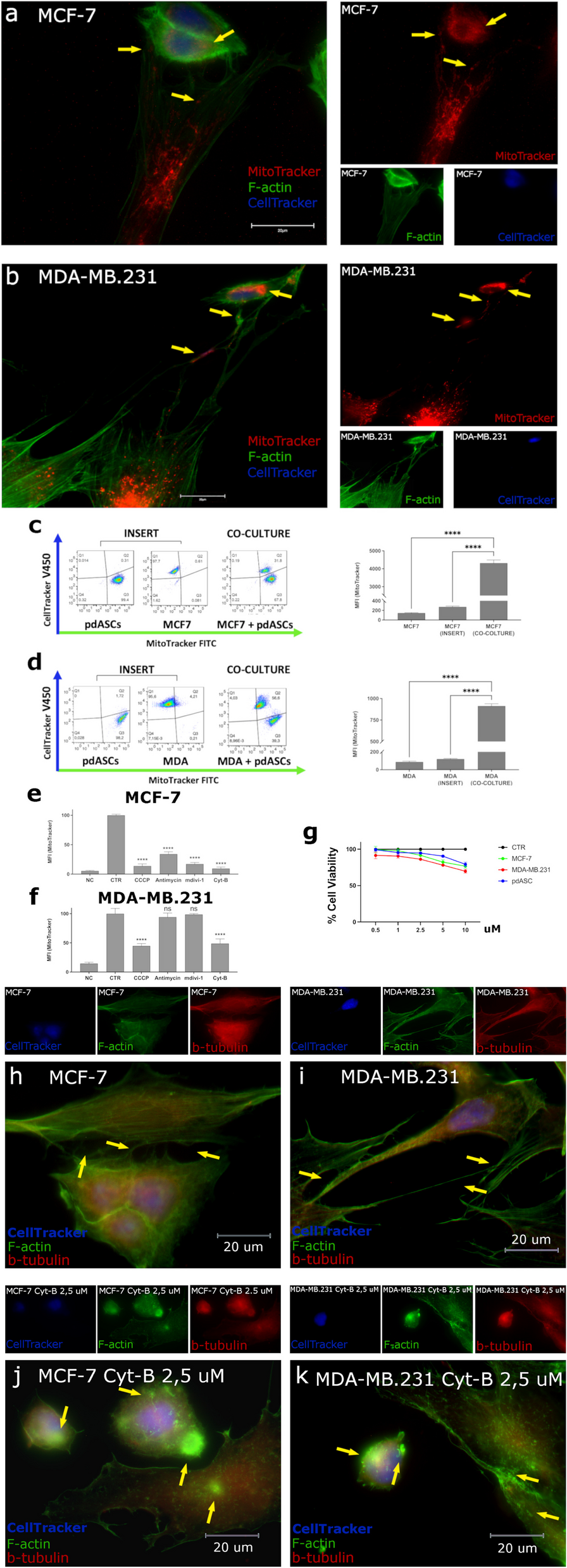
Cells were cultured in 35 mm confocal dishes for 10–12 h, fixed with 4% paraformaldehyde, and stained with 1 µg/ml WGA488 (W11261, Life Technologies, USA) for 15 min (tissue sections were stained with 2 µg/ml for 30 min). Confocal images were acquired using a NIKON A1RSiHD25 laser scanning confocal microscope at 1024 × 1024 pixels. For live-cell imaging, cells were cultured in fibronectin-precoated confocal dishes for 4–6 h before imaging. The cells were maintained at 37 °C with 5% CO2 and monitored using a NIKON A1 microscope at 1024 × 1024 pixels.
Migrasome purification
Migrasome purification was conducted through iodixanol sucrose density-gradient centrifugation using an Optiprep kit (LYSISO1, Sigma-Aldrich). The samples underwent sequential centrifugation steps: 1,000 g for 5 min at 4 °C to eliminate cell bodies, 4,000 g for 20 min at 4 °C to remove cell fragments, and finally 20,000 g for 20 min at 4 °C. The pellet containing the crude migrasome fraction was re-suspended, lysed in extraction buffer (Sigma-Aldrich), and fractionated at 150,000 g for 4 h at 4 °C in a multi-step Optiprep dilution gradient. The gradient included concentrations of 3%, 5%, 8%, 12%, 16%, 19% (sample), 22.5%, and 27%. Fractions were collected and added to 500 µl PBS, followed by centrifugation at 20,000 g for 30 min at 4 °C. The pellet was washed once with PBS, centrifuged at 4 °C, 2,000 g for 10 min, and the supernatant was collected. Further centrifugation at 4 °C, 20,000 g for 30 min resulted in migrasomes for subsequent experiments.
Transmission Electron Microscopy
Cells were cultured in 35 mm dishes precoated with fibronectin (10 mg/ml). After 10–12 h, cells were pre-fixed using a 1:1 ratio of growth medium to 2.5% glutaraldehyde for 5 min at room temperature. Subsequently, cells were fixed with 2.5% glutaraldehyde in PB buffer for 2 h at room temperature, washed three times with PBS, and dehydrated in an ascending gradual series of ethanol (50%, 70%, 90%, 95%, and 100%) for 8 min each. Samples were infiltrated and embedded in SPON12 resin. After polymerisation for 48 h at 60 °C, ultrathin Sect. (70 nm thick) were cut using a diamond knife, picked up with Formvar-coated copper grids (100 mesh), and double-stained with uranyl acetate and lead citrate. Subsequently, air-dried samples were examined using a transmission electron microscope H-7650B at an acceleration voltage of 80 kV.
Transwell invasion assay
Transwell inserts with 8-µm pore size membranes, coated with Matrigel (Corning, 354,277, USA), were allowed to solidify. A total of 1.5 × 10^5 cells, suspended in serum-free medium, were seeded into the upper chambers, while the lower chambers contained medium with 10% FBS as a chemoattractant. After 48 h of incubation, non-invasive cells on the upper membrane surface were removed, and invasive cells on the lower surface were fixed with 4% paraformaldehyde and stained with crystal violet. Images were captured, and migrated cells were quantified using an inverted microscope.
Scratch assay
A scratch assay was utilised to assess cell migration. Cells were seeded in 6-well plates and allowed to reach confluence. A sterile 200-µL pipette tip was used to create a straight scratch across the cell monolayer. After washing with phosphate-buffered saline (PBS) to remove detached cells and debris, images of the scratch were captured immediately after scratching (0 h) and at designated time points during the cell migration process using an inverted microscope. ImageJ software facilitated measuring the scratch width, allowing quantification of cell migration.
Chemotaxis experiments
Chemotaxis experiments were conducted using Transwell inserts with 8-µm pore size membranes. A cell suspension of 1.0 × 10^5 cells in serum-free medium was seeded into the upper chambers, while the lower chamber was filled with medium containing 10% FBS and migrasomes extracted from highly invasive liver cancer cells as a chemoattractant. After a 24-hour incubation, cells on the upper membrane surface were removed with a cotton swab. The invasive cells on the lower surface were fixed with 4% paraformaldehyde and stained with crystal violet. Images were captured, and migrated cells were quantified using an inverted microscope.
Transmission electron microscopy
For transmission electron microscopy, samples were initially pre-fixed using a 1:1 ratio of growth medium to 2.5% glutaraldehyde at 4 °C overnight. Subsequently, samples were fixed with 2.5% glutaraldehyde in PB buffer for 2 h at room temperature, washed three times with PBS, and dehydrated in an ascending gradual series of ethanol (50%, 70%, 90%, 95%, and 100%) for 8 min each. Following infiltration with and embedding in SPON12 resin, 70-nm-thick ultrathin sections were cut using a diamond knife and picked up with Formvar-coated copper grids (100 mesh). Double staining with uranyl acetate and lead citrate was performed, and after air drying, samples were examined with a transmission electron microscope H-7650B at an acceleration voltage of 80 kV.
Field emission scanning electron microscopy
For field emission scanning electron microscopy, samples were fixed with 2.5% glutaraldehyde in PB buffer overnight at 4 °C, washed three times with PB buffer, and post-fixed with 1% osmium containing 1.5% potassium ferrocyanide for 60 min at room temperature. Subsequently, all samples were dehydrated with a graded series of ethanol (50%, 70%, 90%, 95%, and 100%) for 8 min each. After changing ethanol with tert-Butanol, samples were frozen at − 20 °C, then dried with a freeze-drier. The dried samples were coated with an approximately 10-nm-thick gold film by sputter coating before examination with a field emission scanning electron microscope using an SE detector at an acceleration voltage of 3 kV.
ELISA
The concentration of VEGF protein in HCC cell culture medium was determined by ELISA using the Human VEGF Quantikine ELISA Kit (DVE00, R&D Systems) following the manufacturer’s instructions. Absorbance of the samples was determined using the Synergy H4 Hybrid Reader (Biotek, Winooski, VT).
Angiogenesis
Human umbilical vein endothelial cells (HUVECs) were cultured for 24 h and used for the angiogenesis assay. Cells were synchronized by incubating them in ECM containing 0.1% FBS for 12 h. 96-well plates were coated with Matrigel Basement Membrane Matrix (BD Biosciences; diluted in basal ECM at a ratio of 1:1; 20 µl mixture per well) and incubated at 37 °C for 30 min to allow gelation. HUVECs were plated at a density of 5 × 10^4 cells per well. After 4 h of incubation at 37 °C with 5% CO2, pictures were captured with a light microscope.
Tissue microarray and immunohistochemistry
Tissue microarrays were meticulously crafted by Shanghai Zhuoli Biotechnology Co., Ltd (NO. ZL-LVC1801, Zhuoli Biotechnology Co, Shanghai, China), featuring two microarray chips (TMA) housing 90 pairs of tumours and their corresponding adjacent tissues. Acquisition of these samples was carried out with due approval from the hospital. The tissue samples underwent an initial deparaffinization process lasting 1 h, followed by dehydration in ethanol. To quench endogenous peroxidase, a 3% H2O2 treatment was administered. Subsequently, the arrays were subjected to a 20–30 min blocking step using 10% normal goat serum and incubated overnight at 4 °C with primary antibodies (CD151, sc-271,216, Santa Cruz Biotechnology; CD31, 66065-2-Ig, Proteintech). Post PBS washes, the tissue samples were exposed to HRP-conjugated secondary antibody for 1 h at room temperature. Positive signals were visualised using the DAB kit, which reacts with the HRP substrate. Following PBS washes and ethanol dehydration, the tissue slides were meticulously sealed and mounted for microscopic examination.
Statistical analyses
All experiments underwent meticulous repetition three times, and the data is eloquently presented as mean ± standard deviation (SD). Statistical analysis was executed with GraphPad Prism 6.0 software. Group differences were calculated using Student’s t-test or one-way ANOVA with a Tukey post-hoc test. A significance level of P < 0.05 was adopted, and distinctions were considered statistically significant (* P < 0.05, ** P < 0.01, and *** P < 0.001).










留言 (0)