
記住我
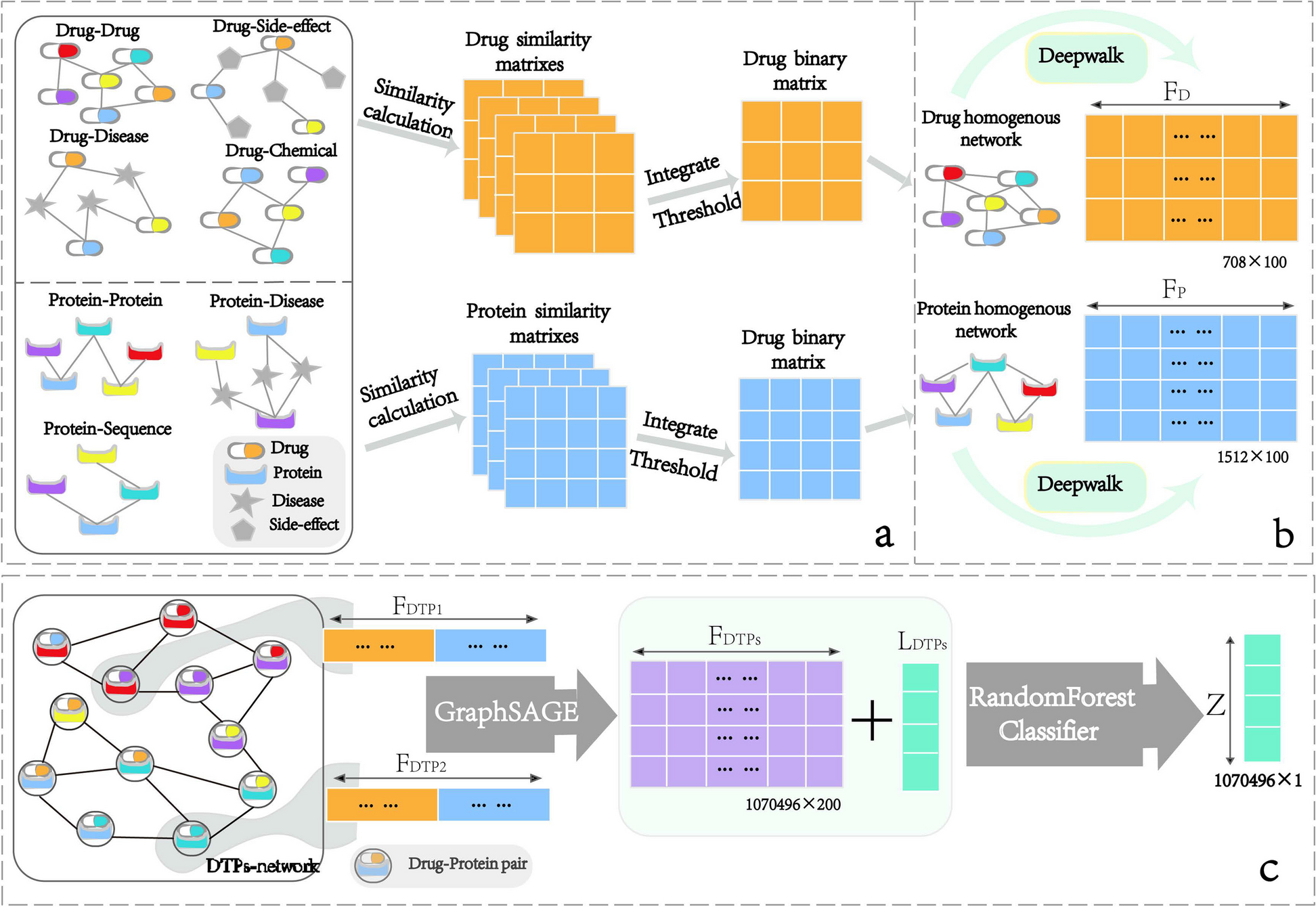


To demonstrate the superiority of DNABERT, we compared it with several typical deep learning models, including Transformer [44], Bert_DPCNN [45], GCN [46], Text_GCN [47], GAT [48], DNN [49], LSTM [50], and GRU [51]. The architectures of these deep learning models were implemented by the DeepBIO server [52] to ensure a fair comparison. As shown in Fig. 2A, in terms of promoter classification, DNABERT outperformed all typical pretrained models and deep learning methods in terms of Sn, Sp, ACC, AUC, and MCC metrics. For example, compared to Transformer, DNABERT achieved a 9.6% improvement in ACC and an 11.7% improvement in MCC. Compared to Bert_DPCNN, DNABERT showed a 3.4% improvement in ACC. Furthermore, compared to GAT, DNABERT demonstrated a 9% improvement in ACC. In terms of promoter strength classification, DNABERT outperformed Transformer by 16.7% in ACC and 33.3% in MCC. Compared to Bert_DPCNN, DNABERT achieved a 15.9% improvement in ACC. Additionally, compared to GAT, DNABERT showed a 22.4% improvement in ACC (refer to the Additional files 2: Table S3 and S4 for detailed data).
Fig. 2
A Comparison of prediction performance of eight baseline predictors in promoter identification and promoter strength prediction. B Performance comparison of ensembles of different base models. The top three figures show the prediction results for promoter identification, while the bottom three figures show the prediction results for promoter strength prediction. C Attention map of four encoding schemes, including 3-mer, 4-mer, 5-mer, and 6-mer, respectively
It is worth noting that compared to other deep learning methods, graph-based deep learning methods generally exhibit poorer performance. This is because DNA sequences often do not contain inherent graph-related information and therefore cannot be properly represented as graphs. This indirectly introduces some noise, which interferes with prediction. On the other hand, common approaches for processing natural language have achieved better performance, such as Transformer, LSTM, and GRU. This is because biological sequences have a great similarity to natural language in essence. Therefore, processing biological sequence data like natural language often yields better results. Among the methods for processing natural language, attention-based methods often achieve better performance due to their powerful ability to understand context. In addition, DNABERT, having been pre-trained on a large amount of biological data, tends to exhibit superior performance in biological scenarios compared to other attention-based methods. In conclusion, these results indicate that DNABERT effectively harnesses the potential of pretrained models. It exhibits superior predictive capability for promoter sequences compared to typical pretrained models and deep learning methods.
Comparison with previously published predictors on benchmark datasetIn order to demonstrate the effectiveness of our model, we compared it with other state-of-the-art predictors on the same benchmark dataset, including Le et al. [30], iPSW (PseDNC-DL) [53], BERT-Promoter [31], iPSW(2L)-PseKNC [34], and iPromoter-CLA [33]. Among the predictors mentioned above, iPSW (PseDNC-DL) and iPSW(2L)-PseKNC focused on solving the problem by utilizing optimal features based on nucleotide compositions. Meanwhile, Le et al. attempted to address the issue by using a combination of fastText model and convolutional neural network (CNN). iPromoter-CLA uses a combination of capsule neural network and recurrent neural network (RNN) to identify promoters and their strengths. In comparison, our model msBERT-Promoter eliminates the need for complex feature encoding projects required by the aforementioned methods. Moreover, most of the above methods use traditional machine learning or deep learning method, without using self-attention mechanism to understand semantic relationship within the sequence. To ensure fairness, we conducted a comparison experiment using the same dataset and evaluation metrics.
Our model outperformed previous models in terms of ACC, Sn, ROC, and MCC for both first-stage promoter recognition and second-stage promoter strength in the independent test dataset. As seen in Table 1, all indicators have achieved good performance, among which achieved a specificity of 0.951, sensitivity of 0.973, accuracy of 0.962, AUC of 0.994, and MCC of 0.923 in the first layer, whereas the second layer achieved the average specificity of 0.786, sensitivity of 0.814, accuracy of 0.798, AUC of 0.874, and MCC of 0.595. These results demonstrated the effectiveness of our proposed model in promoter identification and promoter’s strength classification.
Table 1 Comparison to previously published predictorsIt is noteworthy that the features extracted by deep learning generally outperform traditional handcrafted features, as we have observed in Table 1. In common deep learning methods, utilizing attention mechanisms often leads to better performance. This is one of the reasons why BERT-Promoter and iPromoter-CLA methods outperform previously proposed methods. With the rapid development of large language models, pre-trained models in biological contexts often demonstrate superior performance in biological sequence classification problems. Through unsupervised learning on biological data, these models can enhance their understanding of biological data, thereby exhibiting better performance in downstream tasks related to biology.
Ablation experiment identified the effectivity of msBERT-PromoterFirstly, we conducted ablation experiments to demonstrate the effectiveness of the soft voting ensemble method. We systematically explored all possible combinations and observed that as the number of base models decreased, the performance of these models weakened to varying degrees. As we can see in Fig. 2B, in terms of promoter identification, there was a decrease in model accuracy by 3–14%, AUC by 1–10%, and MCC by 6–26%. For promoter strength prediction, the model’s accuracy decreased by 4–14%, AUC by 2–16%, and MCC by 4–28%. These results indicate that msBERT-Promoter effectively integrates the predictive performance of diverse base models through the soft voting ensemble strategy, resulting in a more robust and high-performing integrated model.
Consequently, in order to validate the efficacy of sequential connectivity in two-stage fine-tuning, we conducted an additional set of experiments. Specifically, we performed fine-tuning on both the promoter identification dataset and the promoter strength prediction dataset separately, denoting them as msBERT-Promoter-X. The experimental outcomes are detailed in Table 2. Notably, in the realm of promoter strength prediction, the predictive performance of msBERT-Promoter surpassed that of msBERT-Promoter-X. This observation underscores the capacity of sequential connectivity to leverage insights acquired during the initial fine-tuning stage to enhance comprehension of the subsequent task, resulting in a 6.63% enhancement in prediction accuracy, a 5.05% increase in AUC, and a notable 12.88% rise in MCC.
Table 2 Ablation study of the two-stage prediction schemeMoreover, to corroborate the validity of the sequence in sequential connectivity, we executed a secondary set of experiments. Initially, we fine-tuned the promoter strength prediction dataset, followed by inputting the promoter identification dataset into the previously fine-tuned model, denoted as msBERT-Promoter-Y. As delineated in Table 2, across both the promoter identification and promoter strength prediction stages, the predictive performance of msBERT-Promoter consistently outperformed that of msBERT-Promoter-Y. This discrepancy can be elucidated from dual perspectives. Primarily, regarding promoter strength prediction, the absence of prior enrichment with promoter identification data hindered the profound understanding of promoter sequence data by msBERT-Promoter-Y, thereby impeding its capability to delve deeply into the task of predicting promoter strength. Subsequently, in the context of promoter identification, the constrained comprehension of promoter data during the initial stage of learning promoter strength prediction tasks in msBERT-Promoter-Y might have engendered negative feedback within its learned experience, potentially hampering its assimilation of promoter identification data and consequently resulting in inferior predictive performance compared to msBERT-Promoter.
In summary, the results derived from this series of experiments underscore the robust rationale and superior performance of our model.
Attention mechanism analysisTo improve the interpretability of the model and pinpoint crucial sequence sites for identifying promoters and predicting their strength, we performed an attention mechanism analysis, visualizing the attention weights of various tokenizer schemes. From Fig. 2C, it can be observed that the high attention weight regions for the four models on the same sample are at positions 1–4, 7–13, 80–81, and 43–51. This indicates that they capture completely different sequence information through different tokenization schemes. Shorter sequence fragments provide the models with a large amount of local information but lack relevant global information. On the contrary, longer sequence fragments grasp broader global information. Different input lengths result in changes in the positions of key features in the encoded sequences, which in turn cause variations in attention distribution.
Therefore, it is crucial to effectively integrate the information extracted from different approaches. To achieve this, we designed several sets of experimental schemes. First is by directly adding all extracted features and then feeding them into a fully connected layer for prediction (called as Ensemble_A). To ensure fairness, all parameters of the fully connected layer are kept consistent with DNABERT’s default parameters. Since directly adding all features may lead to feature redundancy, in the second set of experiments, we incorporated a feature selection algorithm. We used the LightGBM algorithm to rank the added features based on importance and selected 782-dimensional features with importance greater than 0. Subsequently, the selected features were inputted into the fully connected layer for prediction (called as Ensemble_B). As shown in Table 3, experimental results indicate that directly adding features extracted by all base predictors can indeed enhance prediction performance. Moreover, after introducing the feature selection step, the redundancy among features was somewhat alleviated. However, the final performance still did not surpass that achieved using a soft voting ensemble strategy. This is because operations at the feature level often lead to feature redundancy or insufficient feature information. In contrast, the soft voting ensemble strategy integrates at the final prediction level, which avoids compromising the final performance due to feature redundancy, thus demonstrating better performance.
Table 3 Ablation study of the soft voting ensemble scheme on promoter identification and promoter strength predictionIn summary, through attention analysis, we provide insights into the interpretability of the models and emphasize the importance of utilizing a soft voting ensemble strategy to integrate different base learners for improving promoter identification and promoter strength prediction.
The interpretability analysis of soft voting ensemble strategyIn order to gain a more intuitive understanding of how soft voting contributes to improving prediction accuracy, a series of visualizations were implemented. Firstly, an UpSet plot was used to describe in detail the prediction distribution of each base predictor in terms of promoter, non-promoter, strong promoter, and weak promoter. As shown in Fig. 3A, in the first stage (promoter identification stage), 1354 samples were tested, where 345 samples were predicted as promoters and 273 samples were predicted as non-promoters by all four base predictors simultaneously. These samples cannot have their predicted labels changed by the soft voting ensemble strategy; hence, they do not affect the final prediction performance. Additionally, there were 736 samples predicted differently by the base predictors (as promoters or non-promoters), which can potentially improve the final prediction performance through soft voting. Similarly, in the second stage (promoter strength prediction stage), out of 678 tested samples, 65 and 81 samples were predicted as strong promoters or weak promoters by all four base predictors simultaneously. These samples cannot have their final predictions changed by soft voting, but 532 samples predicted differently by the base predictors (as strong promoters or weak promoters) can potentially improve the overall prediction accuracy.
Fig. 3
The interpretability analysis of soft voting ensemble strategy. A The UpSet plot visualizes the intersection of the predictive results from four base models for promoter, non-promoter, strong promoter, and weak promoter. Among them, the horizontal bar chart represents the number of elements contained in different sets, while the vertical bar chart represents the number of elements contained in the intersections of different sets. The black dots connected by black vertical lines indicate which sets are intersecting. B The kernel density estimation plot visualizes the probability distribution of predictions from four base models for promoter, non-promoter, strong promoter, and weak promoter, with the top four representing the first stage and the bottom four representing the second stage
After analyzing that the soft voting strategy has the potential to enhance overall prediction performance, we further explored how the soft voting strategy improves the model’s overall performance by visualizing the kernel density estimates of prediction probabilities for all samples by eight base learners.
As shown in Fig. 3B, in the first stage, the 3-mer, 4-mer, and 5-mer base predictors effectively differentiate between positive and negative samples. The prediction probabilities for positive samples are mainly concentrated in the range of 0.05–0.15, while for negative samples, they are mainly concentrated in the range of 0.95–0.98. This demonstrates the model’s excellent ability to discriminate between different sample classes. However, the 6-mer base predictor does not clearly distinguish between positive and negative samples. The prediction probabilities for positive samples are mainly around 0.47, while for negative samples, they are concentrated in the range of 0.5–0.52 and 0.61–0.63. Despite the 6-mer base predictor showing similar performance to other base predictors, it can be further improved through the soft voting ensemble strategy. By combining the prediction probabilities of the four base predictors and leveraging the strengths of the 3-mer, 4-mer, and 5-mer predictors in terms of prediction probability distribution, the weaknesses of the 6-mer predictor can be compensated for, leading to an 11–14% improvement in promoter identification accuracy. Conversely, although the 3-mer, 4-mer, and 5-mer predictors can effectively differentiate between different sample classes, there are still some samples that are difficult to classify correctly. The 6-mer base predictor can assist in improving the overall prediction performance on these minority samples, demonstrating the collaborative role of each base predictor. In the second stage, all four base predictors can differentiate to a certain degree between positive and negative samples, but there are still some samples that are not properly classified, and the number of misclassified samples is relatively higher compared to the first stage due to the increasing difficulty of the prediction task. However, as shown in the Additional files 3: Table S5 and S6, using the soft voting ensemble strategy in the second stage also results in an 11–14% improvement which further validates the effectiveness and superiority of the soft voting strategy.
t-SNE visualization of extracted featuresTo intuitively compare and analyze how different base predictors extract features from biological sequences, we used t-SNE for visualizing the extracted sequence features. Specifically, we extracted the 768-dimensional features from the twelfth encoding layer of the model and reduced them to two dimensions using t-SNE for easy analysis of how the model classifies sequence data. The experimental results are shown in Fig. 4.
Fig. 4
t-SNE visualization of extracted features by different base predictors. The top four representing the promoter identification stage and the bottom four representing the promoter strength prediction stage
From the visualization, we can clearly see that during the promoter identification stage, all base predictors are able to separate positive samples from negative samples. During the promoter strength prediction stage, the model still overall distinguishes between positive and negative samples, but the proportion of misclassified samples increases. This is due to the increased difficulty of the prediction task. It is worth noting that regardless of the promoter identification or strength prediction stage, the 3-mer base predictor is able to separate all samples into two distinct classes, with a larger distance between samples of different classes. On the other hand, the remaining base predictors only roughly separate samples into two classes, with samples of different classes being closer together. This phenomenon further explains the distribution observed in the kernel density estimation plot in Fig. 3B. Because the 3-mer base predictor can clearly separate all samples into two classes, its predicted probability values are more spread out, and the overlapping region between positive and negative samples is relatively small. In contrast, the other base predictors exhibit samples of different classes being close to each other, which indicates sensitivity to the learned boundary function. Consequently, this leads to a minimal difference in predicted probability values between positive and negative samples, resulting in a larger overlapping region observed between them in the kernel density estimation plot. Therefore, the t-SNE visualization provide further insights into how different base predictors extract and classify sequence features in the stage of promoter identification and strength prediction.
留言 (0)