
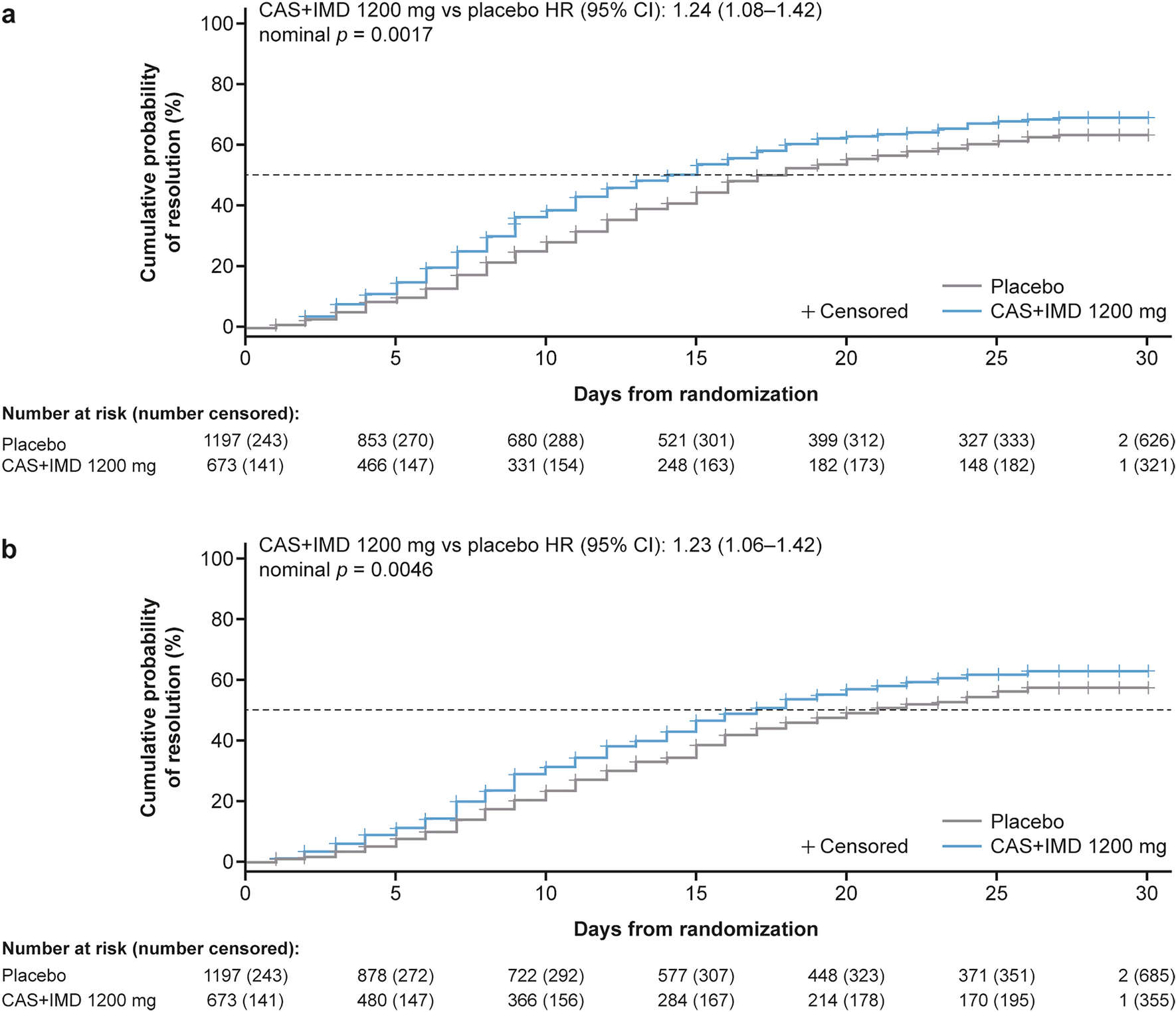
記住我


Data were pooled from three clinical studies (Supplementary Table 1) for which details have been published previously. Study 213725 (NCT03020745) was a phase 1, randomized, double-blind, placebo-controlled, dose-escalation study that assessed the safety, tolerability, and PK of bepirovirsen in healthy volunteers [10]. Participants were randomized 3:1 to receive bepirovirsen 75, 150, 300, or 450 mg (in sequential four-participant single- or multiple-dose cohorts) or placebo, subcutaneously, for up to 3 weeks. Study 205695 (NCT02981602) was a phase 2a, randomized, double-blind, placebo-controlled study that evaluated the efficacy, safety, and PK of bepirovirsen in participants with chronic HBV infection [8]. Participants were randomized 3:1 in each dose cohort to receive bepirovirsen (150 mg for participants Not-on-NAs or 300 mg for participants On-NAs or Not-on-NAs) or placebo subcutaneously for 4 weeks. Study 209668 (B-Clear; NCT04449029) was a phase 2b, multicenter, randomized, investigator-unblinded, parallel group study assessing the efficacy and safety of bepirovirsen in participants with chronic HBV infection either On-NAs or Not-on-NAs [9]. Participants were randomized 3:3:3:1 to receive once weekly: bepirovirsen 300 mg subcutaneously for 24 weeks; bepirovirsen 300 mg subcutaneously for 12 weeks then 150 mg for 12 weeks; bepirovirsen 300 mg subcutaneously for 12 weeks then placebo for 12 weeks; or placebo for 12 weeks then bepirovirsen 300 mg subcutaneously for 12 weeks.
Bepirovirsen concentrations in plasma were assayed using a validated hybridization enzyme-linked immunosorbent assay (ELISA) method (phase 1 study) or liquid–liquid extraction followed by high-performance liquid chromatography with tandem mass spectrometry detection (phase 2 studies). The LLOQ was 1 ng/ml for both methods.
All three studies were reviewed and approved by independent review boards, and written informed consent was obtained from all participants; all studies were conducted in accordance with the International Council on Harmonisation Guideline for Good Clinical Practice and the original principles embodied by the Declaration of Helsinki [8,9,10].
Data Transfer, Assembly, and HandlingPopulation PK analyses were conducted using nonlinear mixed effects modeling (NONMEM software version 7.4.4, ICON plc, Gaithersburg, MD, USA) interfaced with Finch Studio version ≥ 1.1.0 [11] and Perl-speaks-NONMEM version 5.2.6 [12]. SAS version 9.4 (SAS Institute, Inc., Cary, NC) was used for data preparation. R version 4.1.0 and R studio version 1.4.1717 were used for graphical analysis, model diagnostics, and output summaries. Simulations were conducted using mrgsolve (version 0.11.1). Actual dosing times and PK sampling times relative to the first dose were included in the time-ordered dataset, which was used in all analyses. Nominal PK sampling times were used in aggregate data displays where appropriate. Missing baseline covariates were imputed with the median value for continuous variables and with the most common value for discrete variables.
Base Structural Model DevelopmentAn exploratory graphical analysis of observed concentration–time data suggested that bepirovirsen PK is tri-exponential, with dose proportionality, and that participants with chronic HBV infection absorbed bepirovirsen more rapidly than healthy participants (Supplementary Fig. 1). Based on these previous observations and initial modeling attempts, a three-compartment model with the following features was used as the starting base model (Fig. 1): a first-order subcutaneous absorption from a depot compartment, an absorption delay into the central compartment, a distribution to and from the central compartment to two separate peripheral compartments, and linear elimination from the central compartment.
Fig. 1
Schematic of the starting base three-compartment PK model after SC administration. A amount of drug in the compartment, ALAG absorption lag time, CL clearance, CMT compartment, Ka first-order absorption rate constant, PK pharmacokinetic, Q3 apparent intercompartmental clearance between central and shallow peripheral compartments, Q4 apparent intercompartmental clearance between central and deep peripheral compartments, SC subcutaneous, V volume of distribution
Estimated model parameters included first-order absorption rate constant (KA), absorption lag time (ALAG1), apparent clearance following extravascular administration (CL/F), apparent intercompartmental clearance between central and shallow (Q3/F) and central and deep (Q4/F) peripheral compartments, apparent central volume of distribution (V2/F), and apparent volume of distribution of the shallow (V3/F) and deep (V4/F) peripheral compartments.
Baseline body weight was included as covariate effect a priori on CL/F and V2/F, using a power model, based on previous exploratory analyses and initial modeling attempts. Iterations of the base model were explored with the weight effect power model exponents on CL/F and V2/F, both estimated as well as fixed to theoretical values (0.75 and 1 for CL/F and V2/F, respectively). Estimating weight exponents for CL/F and V2/F to be 0.48 and 0.92, respectively, resulted in the best minimum value of the NONMEM objective function (MVOF) and was statistically significantly better than no weight effect or fixed theoretical effect models (Supplementary Table 2).
Interindividual variability (IIV) was estimated for all parameters using exponential error models, assuming log-normal distribution. Residual variability (RV) for plasma bepirovirsen concentration–time data was estimated using a proportional error model and represents a composite of assay variability, intra-subject variability, model misspecification, error in timings of dosing and sampling, participant non-compliance, and other unaccounted factors. The equations used to calculate the IIV and RV are listed in the Supplementary Methods.
Assessment of Covariate EffectsPotential covariate effects were identified by graphical screening prior to stepwise selection. The relationships between random effects or individual error terms (ETAs) and the following pre-specified baseline continuous covariates of interest were analyzed by linear regression and visual inspection of correlation scatter plots for age, weight, serum creatinine, creatinine clearance, albumin, total bilirubin, and alanine aminotransferase (ALT). The relationships between ETAs and the following categorical covariates of interest, disease status (healthy vs. chronic HBV infection), sex, race, NA treatment status, and baseline HBsAg (> 1000 vs. ≤ 1000 IU/ml), were evaluated by visual inspection for differences between groups.
Covariates of clinical interest were included in the covariate search, regardless of whether they demonstrated correlation in the ETA-covariate plots. Analysis was conducted in a stepwise manner; covariates included in the stepwise search are shown in Supplementary Table 3. A univariate forward selection analysis was performed where covariates contributing a change of ≥ 6.635 in the minimum value of the NONMEM objective function (MVOF; level of significance [α] = 0.01, 1 degree of freedom [df]) were considered statistically significant and included in a full covariate model. The stepwise forward selection process was repeated until none of the remaining covariates produced a statistically significant reduction in MVOF. A univariate backward elimination was then performed where covariates contributing an increase of ≥ 10.827 in the MVOF (α = 0.001, 1 df) when removed from the model were considered statistically significant. The most non-significant covariate (highest p value > 0.001) was removed until all remaining covariates were statistically significant.
Model Evaluation and Discrimination CriteriaThe models were assessed using multiple criteria, which were segregated into three groups as follows. The first was evaluation of individual and population mean PK parameter estimates and their precision by % relative standard error (RSE) of the population mean estimate and comparison of estimates to known prior or physiological values. Second was graphical examination of standard diagnostic and population analysis goodness-of-fit plots, including graphical examination of the agreement between the observed and individual post hoc predicted data over time or time since last dose (individual observed and predicted overlays). Finally, the models were also evaluated by measuring the reduction in both RV and IIV, and by comparing the MVOF for nested models.
Model QualificationThe final model was qualified for simulation by performing a dose cohort stratified prediction-corrected visual predictive check (pcVPC). This analysis graphically examined the agreement between the median and the 5th and 95th percentiles of the observed and simulated PK concentrations over time. Based on this graphical analysis, the model was refined as needed to correct for any substantial issues to the fixed or random effects parameters if discordance was seen between the observed and simulated data.
Population PK Model-Based SimulationsThe final population PK model was used to simulate the following bepirovirsen exposure measures at steady state (1000 virtual participants per simulation): Cmax (maximum plasma concentration), Ctrough (trough plasma concentration), and AUCtau (area under the plasma bepirovirsen concentration–time curve during the dosing interval). Measures are presented by dose cohort and other relevant population stratifications including body weight, chronic HBV status, and Asian race versus non-Asian race.
Identification and Exclusion of OutliersAs outliers can negatively impact model convergence and/or final parameter estimates, they were excluded from the PK analysis. Visual inspection of individual and pooled PK data was predominantly used to identify outliers, and additional outliers were detected by graphical exploration of conditional weighted residuals (CWRES) during structural PK model development, and extreme individual post hoc parameter estimates. The final model was rerun after reintroduction of the excluded outliers into the analysis dataset as a sensitivity analysis.
Ethical ApprovalEthical approval was obtained for each study. The studies were conducted in accordance with the International Council on Harmonization Guideline for Good Clinical Practice and the Declaration of Helsinki. The study protocols, any amendments, informed consent, and other information that required pre-approval were reviewed and approved by a national, regional, or investigational center ethics committee or institutional review board, in accordance with the International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice and applicable country-specific requirements, including US 21 Code of Federal Regulations (CFR) 312.3(b) for constitution of independent ethics committees. Each study obtained written informed consent from each participant prior to any study-specific procedures.
留言 (0)