
記住我

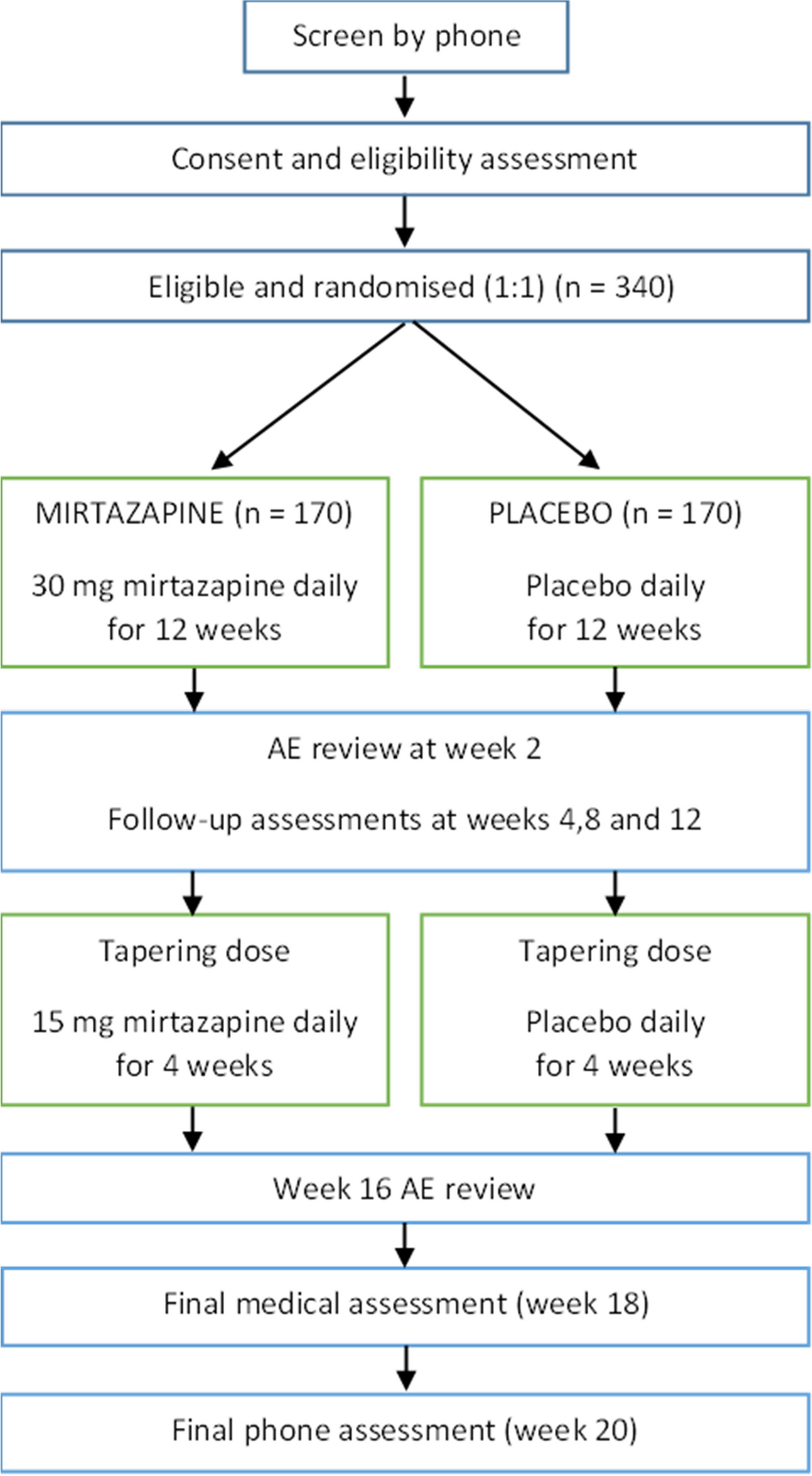




The LIFT trial (efficacy of heeL lIfts For mid-portion Achilles Tendinopathy) will be a parallel group, participant- and assessor-blinded, explanatory, superiority randomised controlled trial with a 12-week follow-up (Fig. 1). Participants will be randomised to a control group (sham heel lift) or an experimental group (heel lifts). To ensure all participants (who will all have some level of pain and disability) receive some form of intervention, both groups will be provided with the same education regarding activity modification. This trial design covers any ethical concerns of not treating participants in pain and will allow the efficacy of heel lifts to be evaluated.
Fig. 1
This trial protocol has been reported in accordance with the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines [17], as well as tendinopathy consensus group reporting recommendations [18]. The SPIRIT checklist is included as Additional file 1. Any changes to the trial protocol will be reported in the final publication and communicated to relevant parties, such as the Ethics Committee, trial registry and participants.
Study settingThe initial assessment will be conducted at a single centre in the Institute for Health and Sport (Victoria University, Melbourne Australia). Publications associated with the trial will be reported according to the Consolidated Standards of Reporting Trials (CONSORT) 2010 [19, 20], and the interventions will be described using the TIDieR checklist [21]. The trial has been registered with the Australian New Zealand Clinical Trials Registry (ACTRN12623000627651).
Ethics approvalEthics approval has been granted from the Monash University Human Ethics Committee (no: 36420). Informed consent will be obtained from all participants (Additional File 2). Ethical standards will adhere to the National Health and Medical Research Council (NHMRC) National Statement [22] and the World Medical Association’s Declaration of Helsinki [23].
Eligibility criteriaParticipants will be recruited by mail-out and emailed advertisements to healthcare practitioners’ in Melbourne. In addition, we will advertise this trial using social media (such as Twitter, Facebook and Instagram), including paid advertisements.
All participants who meet the eligibility criteria will be included. Respondents will initially be screened by telephone interview by a single researcher (JB) to check that they are suitable for the study. Suitable participants will then attend an initial assessment for further eligibility screening. The assessing investigator (JB) is a registered podiatrist with 3 years of experience and will be supported by investigators with expertise in Achilles tendinopathy management and clinical trials (PM: registered physiotherapist; SEM: registered podiatrist).
Inclusion criteria:
Aged 18 to 65 years;
Symptoms of mid-portion Achilles tendinopathy in one or both lower limb(s) for > 6 weeks;
Report maximum Achilles tendon pain severity experienced over the past week that is > 3 out of 10 (using a numerical pain rating scale);
Regularly use footwear that can accommodate heel lifts. This is defined as using footwear that can accommodate heel lifts for at least 8 h per day [24];
Be literate in English and able to complete the questionnaires used in this trial (e.g. VISA-A questionnaire);
Be willing to not receive any treatment on the involved Achilles tendon(s) (other than those allocated in the current study) during the study period;
Be willing and able to attend Victoria University (Melbourne, Australia) on one occasion for assessment.
Mid-portion Achilles tendinopathy will be diagnosed as per the clinical guidelines [7] and musculoskeletal ultrasound [25] using the following criteria:
Report pain in the Achilles tendon during or after weight-bearing activities including walking, running or jumping/hopping;
Pain in the Achilles tendon 2–6 cm proximal to the insertion (as described by the patient and palpated by the investigator);
Gray-scale musculoskeletal ultrasound of the Achilles tendon(s) showing diffuse or local thickening (anterior–posterior) and/or irregular fibre orientation and/or hypoechoic areas within the mid-portion of the Achilles tendon. Certain features are commonly associated with mid-portion Achilles tendinopathy; however, may also exist in asymptomatic individuals [25]. Therefore, if participants exhibit the aforementioned sonographic features accompanied by fluid in the retrocalcaneal bursae (up to 4 mm), focal calcifications, paratenon thickening or calcaneal cortical anomalies (e.g. spurring); they will not be excluded [26].
Exclusion criteria:
Currently pregnant;
Previous Achilles tendon rupture or surgery in the symptomatic lower limb;
Injury or pathology of the lower limb and/or back or any condition that, in the opinion of the investigators, may interfere with participation in the study (e.g. chronic ankle instability);
Concurrent conditions (ankle or other region) that are more severe (pain numerical rating scale) than their worst mid-portion Achilles tendinopathy pain;
Treatment with foot orthoses or heel lifts within the previous 3 months;
Previous breast cancer/and or use of oestrogen inhibitors;
Inflammatory arthritis (e.g. psoriatic arthritis);
Neurological disorders (e.g. Charcot–Marie–Tooth disease);
Taken fluoroquinolone antibiotics within the previous 2 years;
Any injection (e.g. corticosteroid) into the Achilles tendon or surrounding area in the previous 3 months;
Any medical condition that deems a participant unsuitable, based on the opinion of the investigators (e.g. type I or II diabetes).
Baseline assessmentThe baseline assessments were derived from the ICON statement [18]. Participant characteristics (such as age, sex, ethnicity (including if identifies as a First Nations person), education and employment status), smoking status, major medical conditions, number of medications and presentation of tendinopathy (unilateral/bilateral and duration) will be recorded via a custom questionnaire. Height and weight will be measured using a stadiometer and digital scales and body mass index will be calculated as weight (kg)/height (m2). Static foot posture will be assessed using the Foot Posture Index (FPI) [27]. Ankle dorsiflexion range of motion will be assessed with a reliable lunge test technique [28]. Participants’ footwear will be assessed using selected items from the Footwear Assessment Tool [29] and their shoe size documented.
InterventionsRandomisation and blindingParticipants will be randomised to one of two groups: an intervention group (heel lifts) or a control group (sham heel lifts). Both groups will also receive the same guideline recommended education about activity modification. Using Sealed Envelope (https://www.sealedenvelope.com/), participants will be randomised on a 1:1 ratio with random permuted block sizes. Participants will be blinded via limited disclosure, as they will be told that the study will be comparing two types of ‘shoe inserts’. Primary and secondary outcomes will be self-reported; thus, this trial will be assessor blinded. The clinician (JB) administering the interventions will be unable to be blinded. The instructions on using the shoe inserts and education provided will be detailed in a brochure provided to the participants at baseline (Supplementary Material).
Heel lift (intervention group)Participants allocated to the intervention group will be given a pair of commercially available heel lifts (Clearly Adjustable) for bilateral use. The heel lifts are 12 mm in height and made from firm (Shore A 90) multi-layered clear vinyl. To maximise comfort, a 3.2-mm PPT Ultralux top cover will be adhered to the top surface (Fig. 2). The heel lifts will be reduced in 1-mm increments if required (e.g. heel slippage) and the final height will be recorded at baseline. Small, medium and large heel lifts will be available and issued according to the participant’s shoe size. Participants will be shown how to use the heel lifts and asked to wear them for at least 8 h every day. The decision to use these heel lifts were based on the findings of a previous trial that reported this intervention to be safe and more effective for some clinical outcomes than eccentric calf exercises for mid-portion AT over 12 weeks [14].
Fig. 2
Interventions. a Lateral view of heel lift. b Lateral view of sham heel lift. c Dorsal view of interventions (top: heel lift and bottom: sham heel lift)
Sham intervention (control group)Participants allocated to the control group will receive a pair of the sham heel lifts. The following sham intervention may be used; however, we will pilot other interventions (i.e. made of different materials and dimensions) prior to the trial and reserve the right to change the design if required (this will be reported as a change in the protocol on the website trial registration). The intended design of the sham intervention is to not plantarflex the ankle joint but appear as identical to the heel lifts as possible. To achieve this, the sham heel lift will extend the entire length of the shoe and will be made of the same materials as the heel lift (i.e. 1 mm of clear vinyl with a 3.2-mm PPT Ultralux top cover) (Fig. 2). This sham heel lift is necessary in this trial due to participant’s expectation of receiving a ‘take-home’ intervention (i.e. minimises resentful demoralization [30]). The sham intervention will be sized according to shoe size and trimmed (if needed) to fit into the participant’s footwear. Participants will be shown how to use the sham intervention and asked to wear them for at least 8 h every day.
EducationParticipants will be given education regarding the amount of acceptable pain during activity, based on the pain-monitoring model [31]. This approach allows individuals with Achilles tendinopathy to continue with some level of physical activity, while being safe and produces comparable outcomes to complete rest from aggravating activities [31]. Participants will be asked to maintain their regular activities/occupations (rather than complete rest) after receiving their allocated ‘shoe insert’, provided the amount of pain they experience in the Achilles tendon pain does not exceed a score of 5 on a 0–10 pain scale, where 0 is no pain and 10 is worst pain imaginable during exercise/activity. The pain after usual physical activities can reach a 5 on the pain scale but should subside by the following morning. During activity, if the pain in the Achilles tendon exceeds 5 on the pain scale, participants will need to reduce their activity/exercise (if possible).
During the trial, participants will be asked to refrain from using any other treatments for their Achilles tendon pain (besides what they are given in this trial). They will be able to take 500 mg of paracetamol on an ad hoc basis if the tendon(s) is painful and asked to document usage. If participants experience any adverse event (e.g. develop new pain) or have concerns (e.g. uncertainty on using their allocated intervention), they will be encouraged to contact a trial investigator (JB, with support from SM and PM), who will decide if it is safe for the participant to continue with their allocated intervention.
Treatment credibility/expectationThe outcome may be influenced by the participant’s expectations and their beliefs about the credibility of the intervention [32, 33]. Therefore, the credibility of the intervention (participants’ beliefs about the logic underpinning the intervention) and expectancy (participants’ perceptions of how much they may benefit) will be quantified using the Credibility/Expectancy Questionnaire (CEQ) [34]. The CEQ will be administered after the random allocation of the interventions. The CEQ consists of six items that ask participants to rate the credibility of the intervention and their expectancy on a 9-point Likert scale. Higher scores on the scale indicate that the participant considers the intervention to be credible and expects it to be effective. The CEQ is a reliable scale, shown to have good internal consistency and test–retest reliability [34].
OutcomesPrimary and secondary outcome measures [35] will be collected at baseline and at 12 weeks (the endpoint). A summary of the data collection time points for each of the outcome measures is shown in Fig. 3. Follow-up data will be collected using REDCap™ surveys; however, participants will have the option of a postal survey. Where participants have bilateral symptoms, they will be asked to report for their most painful side (or right side, if the Achilles tendons are equally painful) to maintain independence of data [36, 37].
Fig. 3
SPIRIT diagram of enrolment, interventions and assessments for the LIFT trial
Primary outcomeThe primary outcome will be pain intensity at its worst in the previous week using a 11-point numerical rating scale (NRS) with terminal descriptors of ‘no pain’ (score = 0) and ‘worst pain possible’ (score = 10) [38]. This outcome was chosen as it is a core domain in the consensus statement for tendinopathy [35].
Secondary outcomesThe following secondary outcomes will be measured:
1)Pain and disability (measured using the Victorian Institute of Sport Assessment—Achilles (VISA-A)) [39].
2)Global Rating of Change Scale (measured using a 15-point Likert scale ranging from a ‘very great deal worse’ to a ‘very great deal better’). This variable will be dichotomized, with ‘effective’ defined as “somewhat better” or above [40];
3)Function (measured using the Lower Extremity Functional Scale (LEFS) [41]);
4)Health-related quality of life (measured using the VAS component of the EuroQol 5 Dimension 5 Level (EQ-5D-5L) questionnaire [42]);
5)Level of physical activity (measured using the International Physical Activity Questionnaire—short form (IPAQ-SF) [43]).
Other outcomes Co-interventionsThe use of co-interventions to relieve pain at the Achilles tendon will be measured at 4, 8 and 12 weeks via a REDCap survey™. The use of paracetamol rescue medication (number of participants and mean consumption) or any other treatment (e.g. exercise) to manage their Achilles tendon pain.
Adverse eventsAdverse events from the interventions (such as skin blistering or the occurrence of new pain or injuries in other areas of the foot and body) will be assessed at 4, 8 and 12 weeks via a REDCap survey™. Participants will be asked to document the type of adverse event, the body location, the duration and severity of the event [44]. An open-response type format will also be available for participant responses.
AdherenceAdherence will be measured at 4, 8 and 12 weeks via a REDCap survey™. Participants will be asked to provide information regarding the average number of hours per day and number of days they have worn the heel lifts during the preceding 4 weeks.
Biomechanical assessmentAt baseline, a subpopulation of participants who volunteer (n = 40) will undergo a biomechanical assessment to evaluate the acute changes in the biomechanical function of the Achilles tendon as this is the proposed mechanism of action of heel lifts. The variables of interest are Achilles tendon load (ATL), ankle joint moments and plantar/dorsiflexion range of motion (ROM), stance phase duration (SPD), ground reaction force (GRF) and tibial acceleration. One researcher (JB) will perform the assessment, with the support of experts in biomechanical analysis (ST, AG).
After completing a standardised and progressive 7-min warm-up, participants will be asked to walk (5 min) and run (3.5 min) with and without their allocated intervention (i.e. heel lift or sham heel lift) in four testing conditions: (i) walking, shoe only; (ii) walking with the allocated intervention; (iii) running, shoe only and (iv) running with the allocated intervention. The footwear used in testing will be standardised, using the same zero drop Merrell® shoe. The testing sequence will be randomised by a single author (JB). The gait conditions will be fixed at the participant’s optimal walking and preferred running speed to minimise the effect on biomechanical parameters [45].
Optimal walking speed (V) will be calculated to be a fourth of maximal walking speed, using the following calculation: V = sqrt(L × g × 0.25) [46]. The formula is based on the Froude number (0.25), leg length (L) and acceleration of gravity (g). Preferred running speed will be determined using a previously outlined method [47, 48]. Speed will be increased in increments of 0.1 km/h until they first report they have exceeded their preferred ‘comfortable’ running pace. Speed will then be decremented by 0.1 km/h until the participant has confirmed that their ‘comfortable’ running speed has been passed going down. This procedure will be repeated three times and speeds will be averaged to determine the preferred running speed. In between testing, participants will be given as long as required to recover and withdraw if pain exceeds 5 out of 10 on a VAS at any time during the trials [31].
Direct measurement of biomechanical variablesParticipants will walk and run on an instrumented treadmill (Advanced Mechanical Technology Inc., Watertown, MA, USA) that collects GRF at a sampling rate of 1000 Hz. The grade of the contact surface of the treadmill will be maintained in a horizontal position (0%) throughout testing. Three-dimensional kinematic data will be recorded at a sampling rate of 200 Hz from a 14-camera VICON B-10 system (Oxford Metrics Ltd., Oxford, UK). A biomechanical model will be reconstructed from 38 retroreflective markers, placed on proper landmarks of body segments (Additional file 3), which our research team has used previously [49]. Two inertial measurement units (IMUs) will be placed and aligned with the left and right shank respectively to collect tibial acceleration data. Kinematic, IMU and ground reaction force data will be synchronised and collected through Nexus 2.15 software (Vicon Motion Systems Ltd., Oxford, UK).
Achilles tendon load—computed measurementAchilles tendon load (ATL) will be determined using a method described in similar trials [50,51,52]. Achilles tendon forces will be estimated by dividing the ankle plantar flexion moment (PFM) by the Achilles tendon moment arm (MA). The MA will be calculated to 1° increments of ankle flexion (θ) using the following formula [53], which was derived from magnetic resonance imaging [54]: MA = − 0.5910 + 0.08297 × θ − 0.0002606 × θ2. The PFM will be computed around the ankle flexion/extension axis (the axis connecting the medial and lateral malleoli) using Newton–Euler inverse dynamics approach and normalised to body mass (kg) [49].
Sample sizeThe sample size has been determined a priori using G*Power [55] based on pain intensity at its worst in the previous week as the primary outcome measure. We calculated the sample size using an f-test, analysis of covariance (ANCOVA). Using an allocation ratio of 1:1, 90% power, minimal clinically important difference of 1.5 units out of 10 [56], standard deviation of 2.3 [14], moderate effect size [57] (f = 0.33) and a significance level set at a < 0.05, we estimate that a total of 108 participants will be required (99 and an extra 9 for 10% drop-out) [58].
Data monitoringData will be accessible to the trial investigators (JB, SM, AG, ST, PM) and will be stored on a password-protected computer (JB) and uploaded to LabArchives, a secure cloud-based server. This study will have a Trial Management Committee that will be comprised of study investigators (JB, SM, AG, ST, PM). The Committee will meet every 4 weeks to review safety reports, data quality, protocol adherence and retention rates. An independent Data Monitoring and Ethics Committee was considered to not be needed for this trial, as it is relatively short and includes two safe and common interventions [14]. In addition, the participants are not considered to be vulnerable [59, 60]. There will be no formal interim analysis.
Statistical analysisStatistical analysis will be performed using the most recent version of SPSS (IBM Corp., Armonk, NY, USA) available at the time of analysis. Data will be double entered to minimise errors. Demographic data and anthropometric characteristics (e.g. gender, age, foot posture using the FPI, etc.) will be reported by the treatment arm.
The intention-to-treat principle will be used for all participants [61]. Multiple imputation will be used to replace missing data for the primary and secondary outcome measures [62], with the exception being ‘Global Rating of Change Scale, where no data substitution will be applied. Standard tests to assess continuous data for normal distribution will be used and transformation performed if required. The primary outcome measure assessed will be pain intensity (at its worst) at 12 weeks. Continuously scored outcome measures (pain intensity (NRS-11), VISA-A, LEFS, EuroQol 5—VAS, IPAQ-SF and biomechanical outcomes) will be analysed using an ANCOVA with adjustment for baseline scores [63].
Dichotomous data (Global Rating of Change Scale, adverse events and use of co-interventions) will be compared using relative risk, absolute risk increase and number needed to treat or harm. Intervention adherence and CEQ data will be compared between groups using independent t-tests. To complement point estimates, standard deviations, 95% confidence intervals and p-values will be calculated where appropriate.
留言 (0)