Cell culture and establishment of stable cell lines
The human ovarian cancer cell lines used in this paper, SKOV-3, OVCAR8, OVCAR3, OVCA433, HEY, TOV-112D, and Hey-A8 were obtained from ATCC and cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco) with 10% fetal bovine serum (FBS, Gibco) and penicillin (100 U/mL)/streptomycin (0.1 mg/mL) (15140-122, Gibco). For the establishment of stable cell lines, the lentivirus expression plasmids were co-transfected with pxPAX2 and pMD2.G into HEK293T cells. Cells were placed in fresh DMEM for 8 h following transfection. The culture media containing the lentiviral particles were then harvested after 48–60 h of incubation. Lentivirus infection was performed by incubating cells with the virus-containing medium with polybrene for 24 h. Stable cells were then selected on puromycin (1–2 µg/ml). The indicated shRNA sequences are listed in Supplementary Table S1.
Determination of 50% inhibitory concentration
To determine 50% inhibitory concentration (IC50) values of Olaparib (AZD2281; TargetMol, USA), Niraparib (MK-4827; TargetMol, USA), Veliparib (T2591; TargetMol, USA), and Talazoparib (T6523, TargetMol, USA), we measured the cell proliferation rate using Cell Counting Kit-8 (CCK-8) (YEASEN, Shanghai, China). IC50 values were analyzed using GraphPad Prism Version 8.0.
Drug synergy assay
DOT1L inhibitor SGC0946 and PARP inhibitors Olaparib, Niraparib, Veliparib, and Talazoparib were all purchased from TargetMol. For drug synergy studies, ovarian cancer cells (4000/well) were seeded in 96-well plates for overnight incubation and treated with different doses of inhibitors for 5 days. Cell viability was evaluated by measuring the 450 nm absorbance with the Cell Counting Kit-8 (CCK-8) (YEASEN, Shanghai, China). Each concentration was tested in triplicate. The IC50 value was calculated and performed in GraphPad Prism v8.0. Drug synergistic effects were calculated based on the CompuSyn software or SynergyFinder. CI < 1 indicated synergism, CI = 1 indicated additive effects, and CI > 1 indicated antagonism.
Cell apoptosis assay
Treated cells were washed with PBS, digested using trypsin, rinsed in PBS, and then resuspended in 1× binding buffer (YEASEN, Shanghai, China). Cells were incubated with fluorescein isothiocyanate (FITC)-Annexin V (5 µL) for 5 min and 7-AAD (10 µL) for 10 min in the dark at 4 °C. The mixture was further analyzed with a BD Flow Cytometer and the FlowJo software.
Colony formation assay
Cells (2000 cells/well) were plated in 6-well plates as single-cell suspensions, incubated for 24 h, and treated with drugs for 7–14 days. The colonies were fixed with 4% formalin for 15 min and stained with 0.05% crystal violet (Servicebio, Wuhai, China) for 15 min. The number of colonies was counted using the ImageJ 1.52a software.
Western blot
Cells were lysed with RIPA buffer in the presence of a protease inhibitor cocktail (Roche). The protein concentration was determined by a BCA protein assay kit (Wanleibio, Shenyang, China). Equal amounts of proteins were size fractionated by 6-15% SDS-PAGE. gel and transferred into a polyvinylidene difluoride (PVDF) membrane in a wet electron transfer device. 5% skimmed milk in Tris-buffered saline (TBS) containing 0.05% Tween 20 was used to block the membrane for 2 h at room temperature. The blots were incubated with specific antibodies against human primary antibodies, and the signals were detected using horseradish peroxidase-linked anti-mouse or anti-rabbit conjugates as appropriate and visualized using an ECL detection system (GE Healthcare).
Establishment of Olaparib-resistant model
Ovarian cancer cell lines were subjected to a gradual increase in the concentration of Olaparib (from 0.5 to 20 µM) to allow for the development of acquired resistance. Cells with acquired resistance to Olaparib (designated as OlaR) were developed after 3–4 months in drug media. The established Olaparib resistance models were maintained in culture medium with low-concentration Olaparib, and dosing was temporarily ceased prior to conducting experiments.
Antibodies
The antibodies in this study included: H3K79me2 (ab3594, Abcam), Histone H3 (ab1971, Abcam), DOT1L (A300-953 A, Bethyl; sc-390,879, Santa Cruz), PARP1 (13371-1-AP, Proteintech), Flag (F1804, Sigma), P glycoprotein (22336-1-AP, Proteintech), PLCG2 (PTM-6859, PTMBIO), β-tubulin (10068-1-AP, Proteintech), and γ-H2AX (#2577, Cell Signaling Technology).
PARP1‑DNA trapping assay
1 × 106 cells were treated with 10µM Olaparib for 12 h before being harvested for fractionation. The Subcellular Protein Fractionation Kit for Cultured Cells (ThermoFisher Scientific #78,840) was used for cellular fractionation according to the manufacturer’s instructions. Nuclear-soluble and chromatin-bound fractions were then subjected to immunoblotting.
CUT&Tag and data analysis
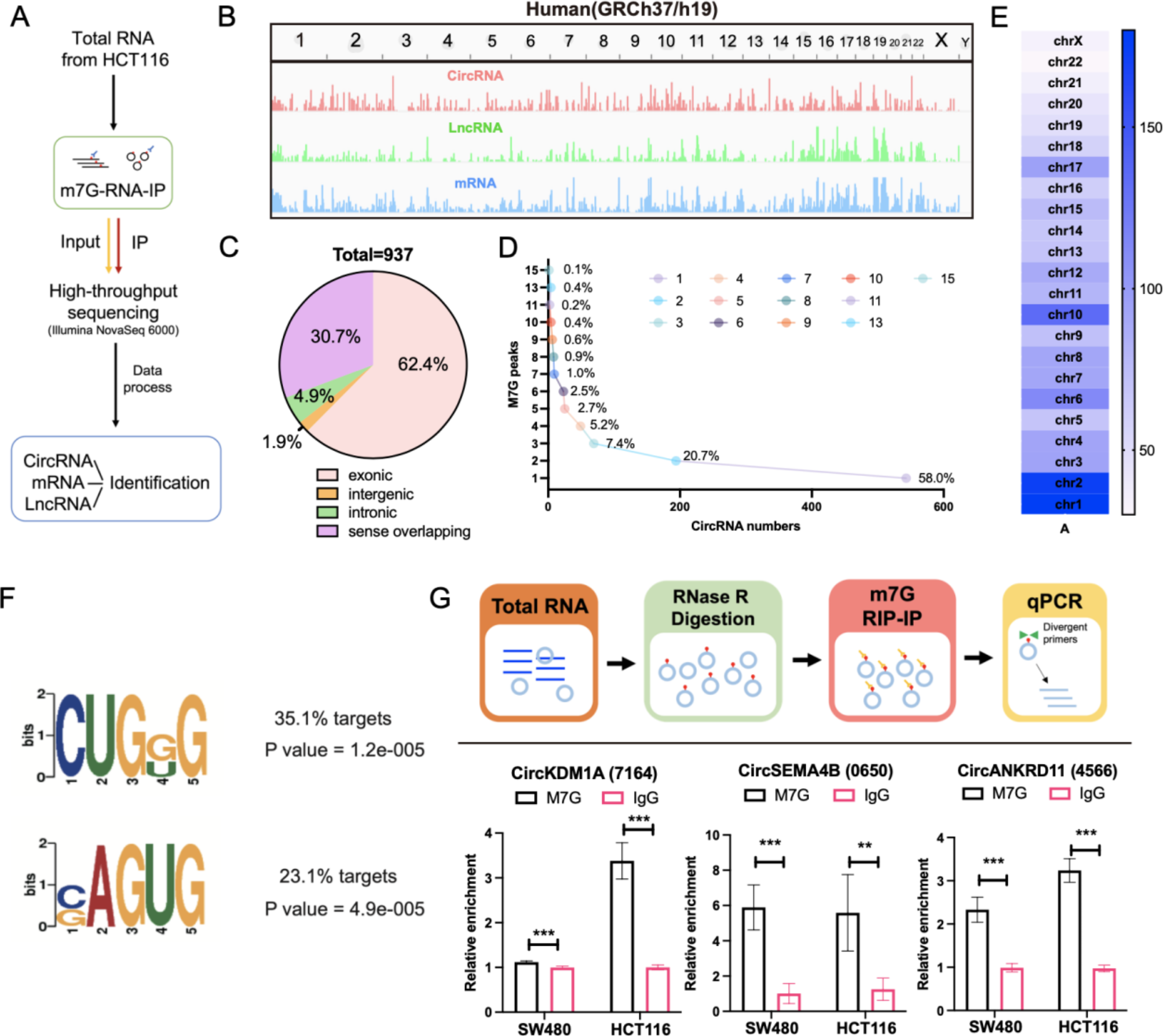
CUT&Tag was performed as previously described [33]. Briefly, 1 × 105 cells were harvested in NE buffer (20 mM HEPES-KOH, pH 7.5, 0.5 mM Spermidine, 10 mM KCl, 0.1% TritonX-100, 10% Glycerol, 1 mM PMSF) and iced for 10 min. ConA beads were pre-washed and resuspended by binding buffer (20 mM HEPES-KOH, pH 7.5, 10 mM KCl, 1 mM CaCl2, 1 mM MnCl2). 10 µl beads were added to each sample and incubated at room temperature for 10 min. The beads were washed with washing buffer (20 mM HEPES-KOH, pH 7.5, 0.5 mM spermidine, 150 mM NaCl, 0.1% BSA) and resuspended in blocking buffer (20 mM HEPES-KOH, pH 7.5, 0.5 mM spermidine, 150 mM NaCl, 0.1% BSA, 2 mM EDTA) at room temperature for 5 min. Primary antibodies (Rabbit monoclonal anti-Histone H3K79me2, CST, 5427 S) were added by 1:100 dilution and incubated at room temperature for 2 h. After being washed with washing buffer, secondary antibodies were added by 1:100 dilution and incubated at room temperature for 30 min. 1.2 µl PA-Tn5 transposomes were added to each sample and incubated at room temperature for 30 min. Beads were resuspended in 30 µl washing buffer with 10 mM MgCl2 and incubated at 37 °C for 1 h. Reactions were stopped by adding 5.5 µl stop buffer (2.25 µL of 0.5 M EDTA, 2.75 µL of 10% SDS and 0.5 µL of 20 mg/ ml Proteinase K) and incubated at 55 °C for 30 min, and then 70 °C for 20 min to inactivate Proteinase K. 0.9X of VAHTS DNA clean beads (VAHTS, Cat. #N411-03) were added to each sample to extract the tagmentated DNA. DNA was purified using phenol-chloroform-isoamyl alcohol extraction and ethanol precipitation. To amplify libraries, 21 µL DNA was mixed with 2 µL of a universal i5 and a uniquely barcoded i7 primer. A volume of 25 µL NEBNext HiFi 2× PCR Master Mix was added and mixed. The sample was placed in a Thermo cycler with a heated lid using the following cycling conditions: 72 °C for 5 min; 98 °C for 30 s; 14 cycles of 98 °C for 10 s and 63 °C for 30 s; final extension at 72 °C for 1 min and hold at 8 °C. The library fragments were purified with XP beads (Beckman Coulter, Beverly, USA). The size distribution of libraries was determined by Agilent 4200 TapeStation analysis, and libraries were mixed to achieve equal representation as desired, aiming for a final concentration as recommended by the manufacturer. Sequencing was performed on the Illumina Novaseq 6000 using 150 bp paired-end following the manufacturer’s instructions.
Raw reads of the fastq format were first processed through in-house scripts. All the downstream analyses were based on high-quality clean data. The clean reads were then aligned to reference genome sequences using the BWA program. The bam file generated by the unique mapped reads as an input file, using the MACS2 software for callpeak with a cutoff q value < 0.05. Peaks were annotated using Homer’s annotatePeaks.pl. Count the results of the annotations and plot the distribution results using R. The Homer’s findMotifsGenome.pl tool was used for Motif analysis.
RNA-seq and data analysis
RNA was harvested from 1 × 106 cells in triplicate and stored in RNAlater RNA stabilization solution (ThermoFisher Scientific). RNA purification, quantification and qualification, library construction and transcriptome sequencing were performed at Jiayin Biotechnology Ltd. (Shanghai, China) according to the manufacturer’s instructions (Illumina, San Diego, CA). Briefly, RNA was isolated using Trizol reagent. mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext. First strand cDNA was synthesized using a random hexamer primer and M-MuLV Reverse Transcriptase (RNase H-). Second strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of the 3’ ends of DNA fragments, NEBNext Adaptor with a hairpin loop structure was ligated to prepare for hybridization. In order to select cDNA fragments of preferentially 250 ~ 300 bp in length, the library fragments were purified with AMPure XP system (Beckman Coulter, Beverly, USA). Then 3 µl USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37 °C for 15 min followed by 5 min at 95 °C before PCR. Then PCR was performed with Phusion High-Fidelity DNA Polymerase, Universal PCR primers and Index (X) Primer. Finally, PCR products were purified (AMPure XP system), and library quality was assessed on the Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Novaseq6000 platform, and 150 bp paired-end reads were generated. After quality control, STAR was used to align clean reads to the reference genome. HTSeq v0.6.0 was used to count the read numbers mapped to each gene. Then the FPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene. We applied the DESeq2 algorithm to filter the differentially expressed genes, after the significant analysis and FDR analysis under the following criteria: (i) |log2FC| > 1; (ii) Pvalue < 0.05.
Patient-derived organoid culture
Patien-tderived organoids (PDOs) were performed as previously described [34, 35]. Upon arrival, ovarian cancer tissues were rinsed in cold PBS with penicillin/streptomycin (GIBCO, 15140-122) for five cycles of five minutes each. Subsequently, the tissues were finely minced into fragments in a sterile dish on ice. Then tissue fragments underwent enzymatic digestion in an 8 mL digestion medium containing 7 mL DMEM medium (GIBCO, C1199500BT), 500 U/mL collagenase IV (Sigma-aldrich, C9407), 1.5 mg/mL collagenase II (Solarbio, C8150), 20 µg/mL hyaluronidase (Solarbio, h8030), 0.1 mg/mL dispase type II (Sigma-aldrich, D4693), 10 µM RHOK inhibitor ly27632 (Sigma-aldrich, Y0503) and 1% fetal bovine serum on an orbital shaker at 37 °C for 30–60 min. Tumor cells were isolated by centrifugation at 300–500 g for 5 min and seeded into Matrigel in a well of pre-warmed 24-well flat bottom cell culture plate (Costar, 3524) and overlayed with 500 µL PDO culture medium after incubation in a 37 °C and 5% CO2 culture incubator for 5–8 min.
The PDO culture medium was refreshed every three days, and PDOs were monitored and photographed at appropriate intervals. Typically, organoids were passaged every 1–2 weeks. For passaging, organoids were gently pipetted out of Matrigel using cold PBS and then mechanically sheared through a 1% BSA-coated pipette tip. Following these steps, the organoids were washed several times with centrifugation at 200–300 g until Matrigel was cleared out. Organoid fragments were suspended in Matrigel and seeded as described above. Cryopreservative medium (serum free) (CELLBANKER™ 2, ZENOAQ, 170,905) was used for organoids cryopreservation. 10 µM RHOK inhibitor ly27632 must be supplemented to the culture medium for organoid resuscitation.
PDO preparation for drug tests
Human ovarian cancer organoids were prepared as previously described. Organoids were harvested and seeded in a 96-well cell culture plate (Corning, 3799) when organoids grew to 50 μm in diameter. The sandwich method was used for drug tests. Before seeding, 50 µL 50% Matrigel (Corning, 356,231, Matrigel mixed with PBS 1:1) was dropped on the bottom of the culture plate as the bottom layer. Then, 10 µL 10% Matrigel (Matrigel mixed with PBS 1:9) containing 50 ± 20 organoids was dropped on the bottom layer as the middle layer. Organoid culture medium (200 µL) was added to each well as the upper layer. Drug tests would start after one day of culturing.
Transcriptional activity
Cells grown to 60–80% confluency were trypsinized and seeded in 24-well culture plates. After 24 h, they were transfected with 500 ng of the indicated DOT1L promoter sequence cloned into the PGL4.0 reporter plasmid. Transfection was performed with Hieff Trans® Liposomal Transfection Reagent (Yeasen, Shanghai, China) following the manufacturer’s instructions. The activities of firefly and Renilla luciferases were measured as relative luminescence units (RLU) using the Dual Luciferase Reporter Assay System (Yeasen, Shanghai, China) 48 h after transfection. Firefly RLU values were normalized to Renilla RLU values and an empty reporter vector. Triplicate samples were systematically included, and experiments were repeated at least three times. The results are shown as mean values with their respective standard deviations.
Tumor formation assay in nude mice
Female BALB/c nude mice aged 4–6 weeks (Shanghai SLAC Laboratory Animal Co., Ltd.; Shanghai, China) were raised in a pathogen-free environment with a 12-h day-night cycle (Department of Laboratory Animal Science in Shanghai Medical College of Fudan University). 100 µL ovarian cancer cells (4 × 106 cells) were subcutaneously injected into the left armpit of each mouse. Olaparib was dissolved in DMSO and diluted to 5 mg/mL with PBS before injection, and 10% DMSO in PBS was used as the vehicle control. When the tumor volumes reached ~ 50mm3, the mice were evenly divided into four groups, and Olaparib (50 mg/kg) and SGC0946 (50 mg/kg) were orally administered to mice three times per week separately or together for 4–6 weeks. The tumor was measured at the indicated time points and was calculated by the formula π/6 × length × width2.
ChIP assay, qPCR and RT-qPCR
A ChIP assay was performed using SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) (9003 S, CST) according to the manufacturer’s instructions. SKOV-3 cells were cross-linked with 1% formaldehyde and then washed with cold PBS, lysed with the lysis buffer, and then sonicated to produce an average DNA length of 500-1,000 bp. Immunoprecipitation was then performed with the indicated antibodies. Purified DNA fragments were analyzed by qPCR using 2×SYBR Green Pro Taq HS Premix (AG11702, Accurate Biology) on a LightCycler 480 Real-Time system (Roche), and precipitated DNA was calculated as a percentage of input DNA. RNA extraction was performed using TRIzol reagent (Life Technologies, Waltham, MA, USA), and cDNA was prepared using Evo M-MLV RT Master Mix Kit (AG11706, Accurate Biology, China). The primers used for the ChIP assay and qPCR are listed in Supplementary Table S1.
Immunohistochemistry (IHC) staining
Totally 273 Chinese patients diagnosed with high-grade serous ovarian carcinoma were involved in this study. All patients had surgical resections at Fudan University Shanghai Cancer Center (FUSCC). Informed consent was obtained from all patients, and the use of clinical samples in this study was approved by the ethics committee of FUSCC. The tumor tissues were fixed with formalin and embedded in paraffin. Following deparaffinization in xylene, rehydration in graded ethanol, and heat-induced antigen retrieval, 4–6-µm-thick tissue sections were incubated with primary antibodies (1:800) at 4 °C overnight, followed by incubation with the corresponding secondary antibodies, visualization using DAB (ZSGB-BIO, Beijing, China), and counterstaining with hematoxylin. The scoring system was based on the staining intensity and extent, as follows: 0 (negative), 1 (weakly positive), 2 (moderately positive), and 3 (strongly positive). The staining positive rate score was calculated as: 1 (0–25%), 2 (26–50%), 3 (51–75%), and 4 (76–100%). The IHC grade was calculated as follows: staining intensity score × positive proportion score. The final score for each sample was the average score for two duplicates. Survival curves were calculated according to the Kaplan-Meier method; survival analysis was performed using the log-rank test.
Quantification and statistical analysis
Statistical analysis was performed using GraphPad Prism v8.0. Statistical significance was determined by the unpaired, two-tailed Student’s t-test. Survival analyses were determined by the Kaplan-Meier curve and log-rank test. All data represent the mean ± SD. P-values were demonstrated in the graphs using * for P < 0.05, ** for P < 0.01, *** for P < 0.001, and **** for P < 0.0001. ns. represents not significant.







留言 (0)