Human tissues sample and databases
Altogether, 15 human normal endometrial tissues and 41 EC tissues were collected from surgical patients at the Union Hospital of Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China) from January 2018 to January 2021. The pathological types of all tissue specimens were confirmed by pathologists. Detailed clinical characteristics and histopathological types of the patients are described in Table 1. All tissue samples were stored in liquid nitrogen until used. Patients read and signed an informed consent form prior to the operation. The Institutional Review Board of Tongji Medical College, Huazhong University of Science and Technology approved the study.
Table 1 Clinicopathologic features of 41 patients with EC and the expressions of circCHD7.Quantitative real-time polymerase chain reaction (qRT-PCR)
According to the instructions, total RNA was extracted from cells or fresh tissues with TRizol (#9108, RNAiso Plus, Takara, Japan). qRT-PCR was performed by Real-Time PCR Kit (Takara, Japan) and the Biosystem StepOne Plus PCR System (ABI). circRNA and mRNA levels were standardized by GAPDH, and RNA expression levels were calculated by the 2-ΔΔCT method. The primers used are listed in Supplementary Table 1.
Cell culture
EC cell lines included Ishikawa, KLE, RL-952, HEC-1A, and HEC-1B, which were purchased from the American Type Culture Collection (ATCC; Manassas, VA). Ishikawa cells were cultured in DMEM/F12 medium (#11330032, Gibco) supplemented with 10% fetal bovine serum (#10099141, Gibco) and 1% streptomycin and penicillin (#PYG0016, Bosterbio, USA). HEB-1C cells were cultured in MEM medium (#41500034, Gibco) with 10% fetal bovine serum (#10099141, Gibco) and 1% streptomycin and penicillin (#PYG0016, Bosterbio, USA). All of them were cultured in the 5% CO2, 37 °C incubator.
Transfection
Lentivirus vector containing short hairpin RNA (shRNA) of circCHD7 were purchased from Genomeditech (Shanghai, China) (Supplementary Table 1). IGF2BP2 three short hairpin RNAs (shRNA), METTL3 short hairpin RNA (shRNA), and negative control shRNA were designed and synthesized by GenChem (Shanghai, China) (Supplementary Table 1). The human PDGFRB cDNA were synthesized by TSINGKE, which were cloned into p3XFLAG-CMV-10 vector (Sigma-Aldrich) to construct overexpression plasmid (Supplementary Table 1). Lentiviral transfection of cells was performed when cell integration reached 20–30%. Adherent cells are fused to about 60% for transfection of plasmids. We established stable cell lines using a lentivirus-mediated delivery system at a multiplicity of infection (MOI) of 30 using polyglutamine. Cells were selected using 2 μg/mL puromycin for 48 h. To construct the stably transfected cell lines, plasmids(2 μg/gel) were transfected into cells by using Lipofectamine 3000 (Life Technologies) according to the manufacturer’s instructions and then the cells were filtered with G418 (Invitrogen) for 4–6 weeks.
RNase R treatment and actinomycin D
2 μg of total RNA was incubated with or without 3 U/μg of RNase for 30 min at 37 °C. The total RNA was obtained for the actinomycin D treatment assay after treating cells with 5 μg/mL of actinomycin D for 0 h, 4 h, 8 h, 12 h and 24 h. After cells were treated with RNAase and actinomycin D, the qRT-PCR procedure was operated as described previously to evaluate changes in expression of circCHD7 and CHD7 mRNA.
Nuclear-cytoplasmic fraction
Isolation of cytoplasmic and nuclear RNA was performed with the Nucleoplasmic RNA Purification Kit (#21000, Norgenbiotek, Canada). Cells were washed twice with pre-cooled PBS, 200 µl of pre-cooled lysis buffer J was added and placed on ice for 5 min. The cell lysate was centrifuged at 12,000 rpm for 10 min at 4 °C to separate the cytoplasmic RNA containing fraction and the nuclear RNA fraction. The cytoplasmic and nuclear solutions were washed sequentially by vortexed with Buffer K and 100% ethanol and subsequently transferred to a centrifuge column for centrifugation. The centrifugation column was washed three times with Washing Solution A and then the RNA was lysed with 50 µl of Elution Buffer E to obtain the RNA solution.
Fluorescence in situ hybridization (FISH) and RNA FISH-immunofluorescence microscopy
Cy3-labeled circCHD7, human U6 and human 18 S probes were designed and synthesized by RiboBioCo. Ltd. (Guangzhou, China) (Supplementary Table 1). FISH assays were conducted by using the RiBoTM Fluorescence in Situ Hybridization Kit (#C10910, RiboBioCo Ltd., Guangzhou, China) according to the instructions. Cells (1 × 10^4 / well) were planted in 24-well plates and cultured overnight in the cell incubator prior to the FISH assay. After incubation in the pre-hybridization solution for 30 min at 37 °C, cells were hybridized with the fluorescent probe overnight at 37 °C shielded from light. The subsequent day, cells were rinsed three times with 4× / 2× / 1× SCC solution for 5 min each followed by DAPI staining for 10 min at 42 °C under light-protected conditions. Cells were eventually observed with confocal microscopy at 400 × magnification.
RNA FISH-immunofluorescence microscopy was to detect co-localization of circCHD7 and IGF2BP2 proteins. Ishikawa cells and HEC-1B cells were fixed, permeabilityed and pre-hybridized. Subsequently, hybridization was performed overnight at 37 °C protected from light using the Cy-3-labelled circCHD7 probe, and the cells were subsequently washed utilizing SSC buffer at 42 °C. Cells were then hatched with primary antibody (1:1000) for 1 h at room temperature, followed by reaction with Alexa Fluor 594 or 488-conjugated secondary antibody (1:1000 dilution; Cell Signaling Technology) and DAPI for 30 min. Images were obtained using confocal microscopy.
Cell counting Kit-8 Assay and colony formation assay
The Cell Counting Kit-8 Assay (#34302, CCK8, Bimake, USA) was used to detect cell proliferation according to the manufacturer’s instructions. After treatment, cells (5 × 10^3 / well) were planted in 96-well plates and incubated for respective time periods (0 h, 24 h, 48 h, 72 h, 96 h). 10 μL of CCK8 solution was added to each well and incubated at 37 °C for 1 h. The absorbance at 450 nm was captured using an automated microplate reader (BioTek, VT, USA).
For the colony formation assay, transfected cells were inoculated into 6-well plates at a density of 600 cells per well and maintained in DMEM/F12 or MEM medium containing 10% FBS for 2 weeks. Cells were washed twice with PBS, fixed in methanol for 10 min and stained with 0.1% crystal violet for 30 min, then colonies were imaged and counted.
EdU assay
Cells (1 × 10^4 cells / well) were transplanted in 96-well plates and incubated overnight at 37 °C with 5% CO2 thermostat prior to EdU (5-ethynyl-2’-deoxyuridine) assay. EdU assays were performed using the EdU kit (#C0071S, Beyotime, shanghai) according to the instructions. EdU solution was prepared as 1:1000 EdU medium. Cells were incubated in pre-prepared 96-well plates with 50 µl / well of EdU medium for 2 h at 37 °C and fixed in 4% paraformaldehyde for 15 min. Cells were incubated with 50 ul/well of click reaction solution for 30 min at room temperature protected from light and then Hochst33342 stained for 10 min. Proliferating cells were imaged with a 200 × microscope and five isolated areas were photographed for counting purposes.
Migration assays
Cell migration assays were performed in 24-well plates. 600 µl of medium containing 20% fetal bovine serum was added to the lower part of the chamber (#3422, Corning, USA) and 200 µl of serum-free medium containing 8 × 10^4 cells were added to the upper part of the chamber. Cells were incubated at 37 °C for 24 h and penetrated from the top to the bottom of the chamber membrane (8 μm pore size). Cells that had penetrated the chamber membrane were fixed in 4% paraformaldehyde for 15 min and stained with 0.1% crystalline violet for 30 min, then were imaged under a 100 × microscope and four separate fields were taken for counting.
Flow cytometry apoptosis assay
Ishikawa and HEC-1B cells were harvested, stained with FITC Annexin V and propidium iodide (PI) (BD Pharmingen) and subsequently analysed for apoptosis using flow cytometry (Becton Dickinson). PI-negative and FITC Annexin V-positive cells were identified at earlier stage of apoptosis, while cells in advanced stage of apoptosis or already dead cells were double positive for PI and FITC annexin V. Results were analyzed with FlowJo software.
RNA pulldown
The biotinylated-circCHD7 probe was synthesized by Sangon Biotech (Shanghai, China) (Supplementary Table 1). The sequence of the circCHD7 antisense probe is complementary to the reverse copy node of circCHD7 only. Briefly, 2 × 107 EC cells were collected, lysed, sonicated, and centrifuged. 50 µl of cell lysate was obtained as input and the remainder was divided equally and mixed with biotin-labelled antisense probes or sense probes and incubated for 2 h at room temperature. The cell lysate containing the probe was then incubated with streptavidin magnetic beads (#HY-K0208, MedChemExpress, USA) for 1 h at room temperature. The magnetic beads were then washed thoroughly with lysis buffer at least three times. The RNA-protein binding mixture was boiled in SDS buffer and the bound proteins were detected by western blot assay and mass spectrometry (MS).
Methylated RNA immunoprecipitation-qPCR (MeRIP-qPCR)
Total RNA was isolated with TRIzol reagent (Invitrogen). methylated RNA immunoprecipitation (MeRIP) was performed using the Magna MeRIP m6A kit (Millipore, Billerica, MA, USA). In brief, an anti-m6A antibody was attached to protein A/G beads and then incubated with fragmented RNA. The enriched RNA was purified and analyzed by qRT-PCR.
RNA immunoprecipitation
RNA-binding protein immunoprecipitation (RIP) assays were performed using the EZ-Magna RIP kit (#17-704, Millipore, Burlington, MA, USA) in accordance with the instructions. The purpose of RIP was to extract and identify protein-bound RNA. Cells (1 × 10^7) were added to the lysis solution and lysed on ice for 5 min. The cells were then centrifuged and the supernatant extracted. After incubation with 5 µg of the target antibody or IgG antibody for 30 min, the magnetic beads were combined with 100 µl of cell lysate supernatant, respectively, and incubated overnight at 4 °C, with rotation. The magnetic bead protein RNA complexes were washed, and then RNA was extracted and subjected to qRT-PCR.
Western blot assays
Total cell or tissue protein was extracted using RIPA lysate (#P0013B, Beyotime, Shanghai, China) and protein concentration was measured using the BCA Protein Assay Kit (#C503021, Singon Biotech, Shanghai, China). Total protein (20 µg) was added to 10% or 12.5% SDS-PAGE gels for gel electrophoresis, followed by transmigration onto polyvinylidene difluoride membranes. After blocking with 5% skimmed milk for 1 h at room temperature, the membranes were incubated with primary antibody overnight at 4 °C. Membranes were washed three times with TBST buffer and incubated with goat anti-rabbit or goat anti-mouse secondary antibodies for 1 h at room temperature. Finally, imaging was performed with The ChemiDoc MP (Bio-Rad, USA). Primary antibodies are listed as follows: IGF2BP2 (1:1000, # 11601-1-AP, Proteintech Group, INC., USA), PDGFRB (1:800, # 13449-1-A, ProteintechGroup, INC., USA), Bcl2 (1:1000, # A0208, ABclonal, Wuhan, China), Bax (1:1000, # A19684, ABclonal, Wuhan, China), METTL3 (1:1000, # A19079, ABclonal, Wuhan, China), STAT3 (1:1000, #9139, Cell Signaling Technology, USA), p-STAT3 (1:1000, #9145, Cell Signaling Technology, USA), JAK2 (1:1000, #3344, Cell Signaling Technology, USA), p-JAK2 (1:1000, #74129, Cell Signaling Technology, USA), GAPDH (1:20000, #AC002, ABclonal, Wuhan, China); secondary antibodies are as follows: HRP Goat Anti-Rabbit IgG (1:8000, #AS014, ABclonal, Wuhan, China), HRP Goat Anti-Mouse IgG (1:8000, #AS003, ABclonal, Wuhan, China), China).
Silver staining and Mass spectrometry analysis
After electrophoresis at 120 V for 75 min, the gel was stained using the Rapid Silver Staining Kit (P0017S, Beyotime) according to the manufacturer’s instructions. Protein profiling was performed by SpecAlly Life Technology Co. (Wuhan, China). In brief, immobilised magnetic bead-bound complexes were washed and digested with sequencing grade modified trypsin. After extraction and purification, peptide samples were identified by mass spectrometry (Q Exactive, Thermo Finnigan, US). The raw mass spectrometry data obtained were retrieved using MaxQuant software (v1.6.2.10). The human reference proteome database of UniProt was used.
RNA-sequence
To explore in more depth how circCHD7 regulates gene expression in EC cells, RNA-seq was performed in Ishikawa cell cells stably expressing circCHD7. Total RNA was isolated from cultured cells using TRIzol and then sequenced using the NovaSeq 6000 platform (Illumina, USA). RNA-seq was performed as previously reported [26]. KEGG pathway analysis was performed on differentially expressed genes from RNA-seq (|log2(FC) | > 2, P < 0.05).
Hematoxylin-eosin staining
Dewaxing: xylene I and II for 10 min apiece. Dehydration: 100% (I and II), 90%, 80% and 70% alcohol for 5 min each and rinsed 3 times under running water for 5 min each. Staining: sections were stained for 5 min using hematoxylin and rinsed under running water. Acetic acid fractionation for 1 min and rinsing of sections under flowing water. Eosin staining for 1 min and rinsing the slides under running water. 70%, 80%, 90%, and 100% alcohol for 10 s each and xylene for 1 min to dehydrate the slides. Slides were dried spontaneously and sealed with drops of neutral glue.
Immunohistochemistry (IHC)
Paraffin sections of tumour tissue were dewaxed and dehydrated, hydrated and repaired with citric acid. After peroxidase blockade, tissue sections were incubated overnight at 4 °C with primary antibody. Slides were washed with PBS and incubated with secondary antibody for 30 min at 37 °C. Sections were finally sealed and observed with the microscope. Staining intensity was categorized as negative (score = 0), weak (score = 1), moderate (score = 2) and strong (score = 3); the quantities of positively stained cells were 0–5%, 5–25%, 26–50%, 51–75% and 76–100% respectively. The ultimate staining score was the multiplication of staining intensity and the number of positively stained cell products. Antibodies: IGF2BP2 (1:2000, # 11601-1-AP, Proteintech Group, INC., USA), PDGFRB (1:1000, # 13449-1-A, Proteintech Group, INC., USA), Bcl2 (1:1000, # A0208, ABclonal, Wuhan, China), Bax (1:1000, # A19684, ABclonal, Wuhan, China).
In vivo tumor xenografts
Animal experiments were supported and approved by the Animal Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology (NO.3722). 6-week-old female BALB/c nude mice were purchased from ShuLaiBao Biotechnology Company (Wuhan, China) and Ishikawa cells (4 × 10^6 per mouse) were injected subcutaneously into the back of the mice, after random grouping (6 per group) to conform to a normal or approximately normal distribution. Mice were housed in specific pathogen-free animal houses for 28 days. The subcutaneous tumour sizes in the mice were measured each week. At the end of the experiment (after 4 weeks), tumour sizes were measured and the mice were sacrificed, the tumours removed and weighed. The tumour volume was calculated as: a × b^2, where a represented the longest diameter of the tumour and b was the length of the diameter perpendicular to a. For animal studies, the operation should obey blinding.
Statistical analysis
All statistical analyses were performed with GraphPad Prism v. 8.01 (GraphPad Software, La Jolla, CA, USA). Student’s t-test was applied to compare the values of the test and control groups. For multiple comparisons, differences were analyzed by one-way ANOVA and Turkish post hoc test. p < 0.05 was considered statistically significant (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). The variance is similar between the groups that are being statistically compared.

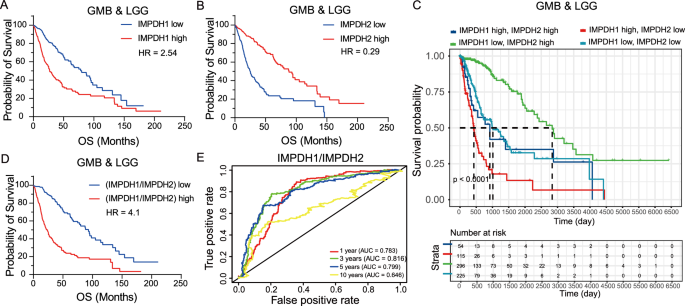
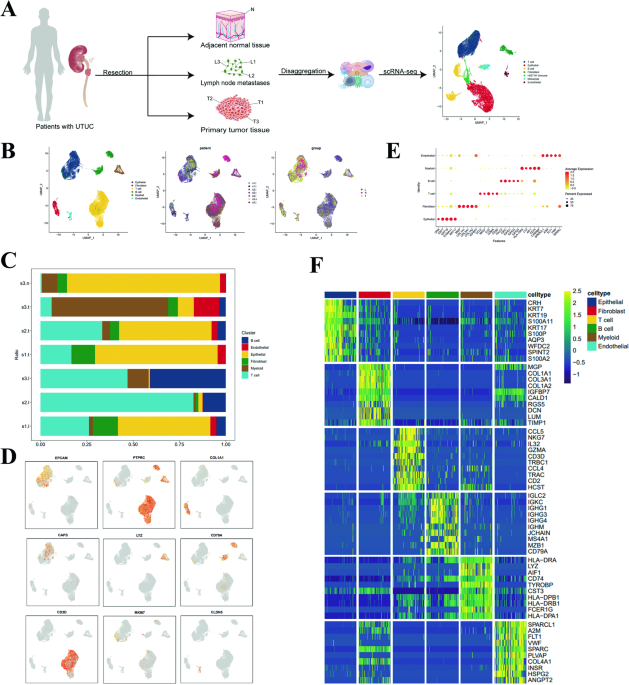
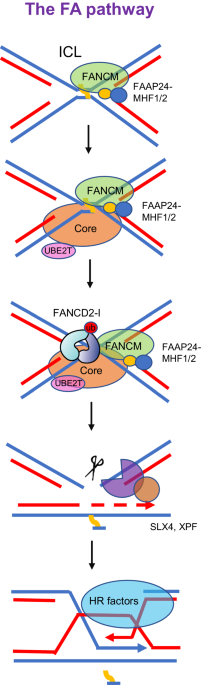
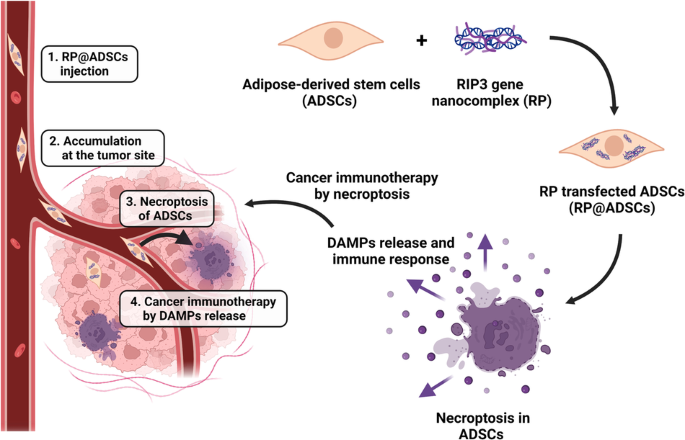
留言 (0)