
記住我
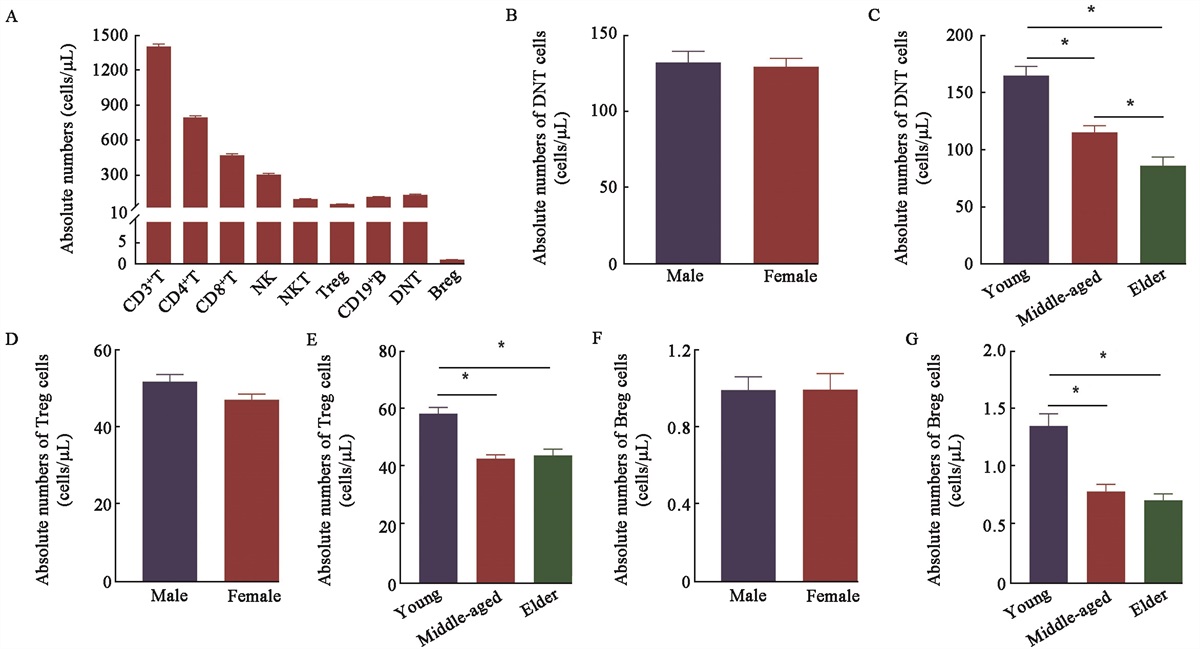








To the Editor: Chronic obstructive pulmonary disease (COPD) is a global public health challenge without effective treatment currently available, affecting >300 million individuals worldwide. Comprehensively understanding the pathogenesis of this disease is required. Recently, ferroptosis, a form of cell death driven by iron-dependent lipid peroxidation, has been associated with the pathogenesis of COPD.[1] Multiple signaling pathways, such as glutathione peroxidase 4 (GPX4), ferroptosis suppressor protein 1 (FSP1), and tetrahydrobiopterin-dependent pathways, as well as iron metabolism, are involved in regulating ferroptosis[2,3] [Figure 1A]. Here, we provide a comprehensive overview of recent advances in understanding the role of ferroptosis in COPD, highlighting potential therapeutic strategies targeting ferroptosis for treating the disease.
 Figure 1:
Figure 1: (A) Signaling pathways involved in regulating ferroptosis. (B–D) Ferroptosis in BECs (B), macrophages (C), and ECs (D) contributes to COPD. (E) Targeting ferroptosis in various pulmonary cells to mitigate COPD. α-TOH: α-tocopherol; ACSL4: Acyl-CoA synthetase long-chain family member 4; ALOX15: Arachidonate 15-lipoxygenase; ALOX5: Arachidonate 5-lipoxygenase; BECs: Bronchial epithelial cells; COPD: Chronic obstructive pulmonary disease; CoQ10(H): Oxidized form of coenzyme Q10; CoQ10H2: Reduced form of coenzyme Q10; circSAV1: Circular RNA SAV1; DHFR: Dihydrofolate reductase; ECs: Endothelial cells; FSP1: Ferroptosis suppressor protein 1; FTH: Ferritin heavy chain; FTL: Ferritin light chain; GPX4: Glutathione peroxidase 4; GSH: Glutathione; GSS: Glutathione synthetase; GSSG: Oxidized glutathione; GSR: Glutathione-disulfide reductase; H2S: Hydrogen sulfide; HMOX-1: Heme oxygenase-1; iNOS: Inducible nitric oxide synthase; IREB2: Iron-responsive element binding protein 2; LOX: Lipoxygenase; LTB4: Leukotriene B4; MFG-E8: Milk fat globule-EGF factor 8; MMP9: Matrix metalloproteinase 9; MMP12: Matrix metalloproteinase 12; NAD(P)H: Nicotinamide adenine dinucleotide phosphate; NCOA4: Nuclear receptor coactivator 4; Nrf2: Nuclear factor erythroid 2-related factor 2; PGE2: Prostaglandin E 2; PLOO•: Lipid peroxyl radicals; PLOOH: Phospholipid hydroperoxides; PRMT7: Arginine methyltransferase 7; PTGS2: Prostaglandin E synthase 2; PUFAs: Polyunsaturated fatty acids; PUFA-PLs: PUFA containing phospholipids; RAP1A: Ras-related protein 1A; SIRT3: Sirtuin 3; SLC40A1: Solute carrier family 40A1; STEAP3: Six-transmembrane epithelial antigen of prostate 3; TF: Transferrin; TFRC: Transferrin receptor; TXNRD1: Thioredoxin reductase 1; USP14: Ubiquitin-dpecific protease 14; VK: Vitamin K; VKH2: Vitamin K hydroquinone; YTHDF1: YTH N6-methyladenosine RNA binding protein 1; Znpp: Zinc protoporphyrin.
Ferroptosis in bronchial epithelial cells (BECs) contributes to COPD: Excessive iron deposition in cigarette smoke (CS)-exposed BECs leads to increased ferritin expression. This process activates ferritinophagy mediated by nuclear receptor coactivator 4 (NCOA4). Notably, the knockdown of NCOA4 in mice reduces CS-induced ferroptosis.[4] In COPD, downregulated nuclear factor erythroid 2-related factor 2 (Nrf2) expression is associated with Nrf2 promoter hypermethylation. Conversely, increased Nrf2 expression enhances GPX4 levels, inhibits ferroptosis in BECs, and mitigates COPD.[5] Moreover, CS extract (CSE)-induced lipid peroxidation leads to sirtuin3 inactivation in COPD, promoting inducible nitric oxide synthase (iNOS) levels and potentially triggering ferroptosis in BECs in COPD.[6] Milk fat globule epidermal growth factor 8 (MFG-E8) has been identified as a mitigator of CSE-induced ferroptosis in BECs, and ubiquitin specific peptidase 14 (USP14) stabilizes MFG-E8 protein, suppressing CS-induced ferroptosis.[7] m6A-modified circular RNA SAV1 (circSAV1) forms an RNA–protein complex that promotes the translation of iron-responsive element binding protein 2 (IREB2) mRNA. Increased IREB2 levels disrupt iron homeostasis, resulting in the accumulation of a labile iron pool and lipid peroxidation, contributing to ferroptosis in epithelial cells. Notably, the knockdown of circSAV1 reversed CSE-induced ferroptosis, suggesting its crucial regulatory role in this process.[8] Overall, ferroptosis in BECs contributes to COPD [Figure 1B].
Ferroptosis in macrophages contributes to COPD: Lipid peroxidation accumulation in COPD activates ferroptosis-sensitive M2-like macrophages.[9] Protein arginine methyltransferase 7 (PRMT7) regulated by nuclear factor kappa B (NF-κB)/RelA activation expression is also associated with lung injury severity in COPD. Pro-inflammatory macrophages with increased PRMT7 expression stimulate arachidonate 5-lipoxygenase and leukotriene B4 release, inducing acetyl coenzyme A (acyl-CoA) synthetase long-chain family member 4 (ACSL4) expression in BECs, rendering them more susceptible to ferroptosis in COPD.[1,10] Moreover, increased M2 macrophages and levels of matrix metalloproteinase (MMP) 9 and MMP12 were observed in CS-exposed mice and macrophages co-cultured with CSE-treated BECs.[11] CS exposure led to elevated NCOA4 levels and increased ferritin in BECs, resulting in iron overload and lipid peroxidation-induced ferroptosis. This ferroptosis in BECs may contribute to macrophage M2 polarization in COPD. Overall, ferroptosis in macrophages contributes to COPD [Figure 1C].
Ferroptosis in endothelial cells (ECs) may disrupt the interaction between ECs and BECs, exacerbating COPD: Wang and Xia[12] observed ferroptosis in ECs exposed to CSE and homocysteine, characterized by reduced GPX4 and FSP1 expression and increased ACSL4 expression. This suggests that ferroptosis in ECs contributes to the disruption of the pulmonary microvascular barrier in COPD. Notably, Yu et al[13] discovered that normal ECs release exosomes containing micro RNA (miR)-26a-5p, which can be transported into BECs. Within the BECs, miR-26a-5p targets prostaglandin-endoperoxide synthase 2 (PTGS2) mRNA, potentially inhibiting ferroptosis. These findings suggest that ferroptosis in ECs may disrupt the beneficial interaction between ECs and BECs, exacerbating COPD [Figure 1D].
Targeting ferroptosis in BECs mitigates COPD: Dihydroquercetin activates Nrf2-mediated pathways to inhibit CSE-induced ferroptosis in BECs in COPD.[14] Curcumin also attenuates CSE-induced inflammation and ferroptosis in BECs.[15] Scutellarein is a potent inhibitor of ferroptosis through chelating Fe2+ and reducing intracellular Fe2+ levels, but its antiferroptotic effects can be hindered by arachidonate 15-lipoxygenase overexpression.[16] In addition to these natural compounds, treatment with deferoxamine, ferrostatin-1, and liproxstatin-1 can inhibit CSE-induced ferroptosis in BECs.[4,8] Sodium pyruvate activates the GPX4/Nrf2 pathway to inhibit CSE-induced ferroptosis in BECs.[17] Hydrogen sulfide (H2S) activates Nrf2 and peroxisome proliferator-activated receptor-γ signaling pathways, regulating NCOA4-mediated ferritin autophagy.[18] Sodium hydrosulfide (NaHS), a H2S donor, mitigates lipid peroxidation and ferroptosis in BECs. Furthermore, gene therapy targeting ACSL4 using a lentiviral system (si_ACSL4) reduces ACSL4 expression and inhibits ferroptosis in BECs, showing promise as a strategy for treating COPD.[12] Overall, targeting ferroptosis in BECs mitigates COPD [Figure 1E].
Targeting ferroptosis in macrophages mitigates COPD: A study using CSE to induce ferroptosis in macrophages found that M1 macrophages were more resistant to CS-induced ferroptosis, whereas M2 macrophages were more susceptible.[9] M2 macrophages exhibited higher levels of heme oxygenase-1 (HMOX1) and iron than M1 macrophages. Inhibiting HMOX1 using zinc protoporphyrin (ZnPP) effectively prevented macrophage ferroptosis induced by CSE. Moreover, treatment with ferrostatin-1, ZnPP, and deferoxamine reduced Fe2+ levels in bronchoalveolar lavage fluid and the release of ferritin from macrophages. These treatments protected against ferroptosis in macrophages in COPD.[9] Overall, targeting ferroptosis in macrophages mitigates COPD [Figure 1E].
Targeting ferroptosis in ECs mitigates COPD: CS exposure exacerbates dyslipidemia, promotes atherosclerotic plaque formation, induces lung function decline, causes pathological damage, disrupts pulmonary microvascular barrier function, and triggers ferroptosis in ECs in mice fed a high-fat diet. By mitigating ferroptosis and preserving pulmonary microvascular barrier function, Tongxinluo treatment can prevent COPD complicated with atherosclerosis.[19] Overall, these findings highlight the potential of targeting ferroptosis in ECs as a therapeutic approach for COPD [Figure 1E].
In summary, we comprehensively analyze the role of ferroptosis in BECs, macrophages, and ECs in COPD. Moreover, we highlight potential therapeutic strategies targeting ferroptosis in these cells for COPD treatment. However, several important questions remain to be addressed. First, future studies should use integrative omics approaches to elucidate the underlying mechanisms of ferroptosis in COPD. Second, in addition to BECs and macrophages, further investigation is required to understand the effects of ferroptosis inhibitors on other pulmonary cell types in COPD. Finally, it is crucial to determine whether ferroptosis occurs during the entire development process of COPD or it mainly manifests in advanced stages.
FundingThis work was supported by the National Natural Science Foundation of China (No. 82200084), the Natural Science Foundation of Sichuan Province (No. 2023NSFSC1456), the Postdoctoral Science Foundation funded project of Sichuan Province (No. TB2023047), the Fundamental Research Funds for the Central Universities, and the Sichuan University Postdoctoral Interdisciplinary Innovation Fund (No. 0020404153020).
Conflicts of interestNone.
References 1. Günes Günsel G, Conlon TM, Jeridi A, Kim R, Ertüz Z, Lang NJ, et al. The arginine methyltransferase PRMT7 promotes extravasation of monocytes resulting in tissue injury in COPD. Nat Commun 2022;13:1303. doi: 10.1038/s41467-022-28809-4. 2. Xu J, Zhou F, Wang X, Mo C. Role of ferroptosis in pregnancy related diseases and its therapeutic potential. Front Cell Dev Biol 2023;11:1083838. doi: 10.3389/fcell.2023.1083838. 3. Lyu T, Li X, Song Y. Ferroptosis in acute leukemia. Chin Med J 2023;136:886–898. doi: 10.1097/CM9.0000000000002642. 4. Yoshida M, Minagawa S, Araya J, Sakamoto T, Hara H, Tsubouchi K, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun 2019;10:3145. doi: 10.1038/s41467-019-10991-7. 5. Zhang Z, Fu C, Liu J, Sai X, Qin C, Di T, et al. Hypermethylation of the Nrf2 promoter induces ferroptosis by inhibiting the Nrf2-GPX4 axis in COPD. Int J Chron Obstruct Pulmon Dis 2021;16:3347–3362. doi: 10.2147/copd.S340113. 6. Zi Y, Wang X, Zi Y, Yu H, Lan Y, Fan Y, et al. Cigarette smoke induces the ROS accumulation and iNOS activation through deactivation of Nrf-2/SIRT3 axis to mediate the human bronchial epithelium ferroptosis. Free Radic Biol Med 2023;200:73–86. doi: 10.1016/j.freeradbiomed.2023.03.002. 7. Cui Y, Luo L, Zeng Z, Liu X, Li T, He X, et al. MFG-E8 stabilized by deubiquitinase USP14 suppresses cigarette smoke-induced ferroptosis in bronchial epithelial cells. Cell Death Dis 2023;14:2. doi: 10.1038/s41419-022-05455-8. 8. Xia H, Wu Y, Zhao J, Cheng C, Lin J, Yang Y, et al. N6-methyladenosine-modified circSAV1 triggers ferroptosis in COPD through recruiting YTHDF1 to facilitate the translation of IREB2. Cell Death Differ 2023;30:1293–1304. doi: 10.1038/s41418-023-01138-9. 9. Li Y, Yang Y, Guo T, Weng C, Yang Y, Wang Z, et al. Heme oxygenase-1 determines the cell fate of ferroptotic death of alveolar macrophages in COPD. Front Immunol 2023;14:1162087. doi: 10.3389/fimmu.2023.1162087. 10. Ding K, Liu C, Li L, Yang M, Jiang N, Luo S, et al. Acyl-CoA synthase ACSL4: an essential target in ferroptosis and fatty acid metabolism. Chin Med J 2023;136:2521–2537. doi: 10.1097/CM9.0000000000002533. 11. Liu J, Zhang Z, Yang Y, Di T, Wu Y, Bian T. NCOA4-mediated ferroptosis in bronchial epithelial cells promotes macrophage M2 polarization in COPD emphysema. Int J Chron Obstruct Pulmon Dis 2022;17:667–681. doi: 10.2147/copd.S354896. 12. Wang Y, Xia S. Relationship between ACSL4-mediated ferroptosis and chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2023;18:99–111. doi: 10.2147/copd.S391129. 13. Yu T, Wang P, Wu Y, Zhong J, Chen Q, Wang D, et al. MiR-26a reduces inflammatory responses via inhibition of PGE2 production by targeting COX-2. Inflammation 2022;45:1484–1495. doi: 10.1007/s10753-022-01631-2. 14. Liu X, Ma Y, Luo L, Zong D, Li H, Zeng Z, et al. Dihydroquercetin suppresses cigarette smoke induced ferroptosis in the pathogenesis of chronic obstructive pulmonary disease by activating Nrf2-mediated pathway. Phytomedicine 2022;96:153894. doi: 10.1016/j.phymed.2021.153894. 15. Tang X, Li Z, Yu Z, Li J, Zhang J, Wan N, et al. Effect of curcumin on lung epithelial injury and ferroptosis induced by cigarette smoke. Human Exp Toxicol 2021;40:S753–S762. doi: 10.1177/09603271211059497. 16. Liu L, Zhang Y, Wang L, Liu Y, Chen H, Hu Q, et al. Scutellarein alleviates chronic obstructive pulmonary disease through inhibition of ferroptosis by chelating iron and interacting with arachidonate 15-lipoxygenase. Phytother Res 2023;37:4587–4606. doi: 10.1002/ptr.7928. 17. Zhao Z, Xu Z, Chang J, He L, Zhang Z, Song X, et al. Sodium pyruvate exerts protective effects against cigarette smoke extract-induced ferroptosis in alveolar and bronchial epithelial cells through the GPX4/Nrf2 axis. J Inflamm (Lond) 2023;20:28. doi: 10.1186/s12950-023-00347-w. 18. Wang Y, Liao S, Pan Z, Jiang S, Fan J, Yu S, et al. Hydrogen sulfide alleviates particulate matter-induced emphysema and airway inflammation by suppressing ferroptosis. Free Radic Biol Med 2022;186:1–16. doi: 10.1016/j.freeradbiomed.2022.04.014. 19. Wang Y, Kuang X, Yin Y, Han N, Chang L, Wang H, et al. Tongxinluo prevents chronic obstructive pulmonary disease complicated with atherosclerosis by inhibiting ferroptosis and protecting against pulmonary microvascular barrier dysfunction. Biomed Pharmacother 2022;145:112367. doi: 10.1016/j.biopha.2021.112367.
留言 (0)