
記住我



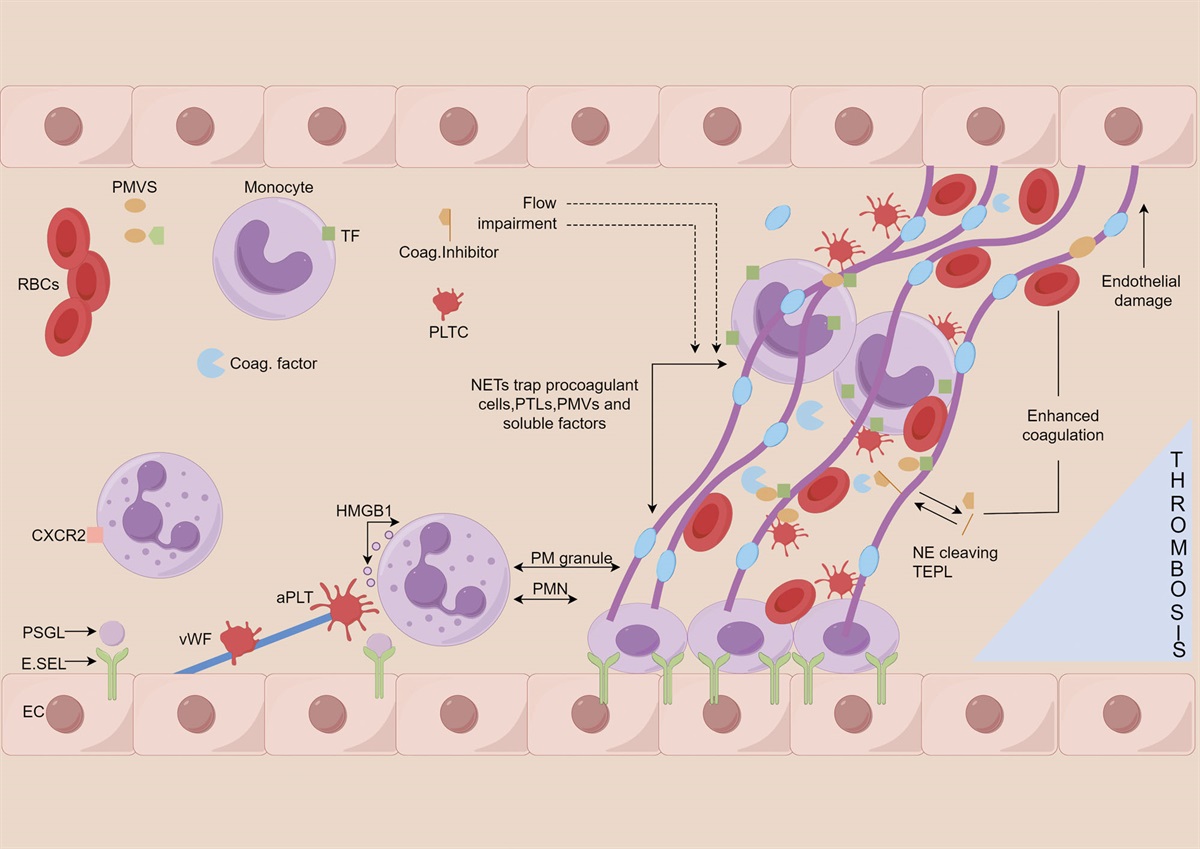



Inflammation is an immune response of the body to various stimuli and protects and repairs damaged tissues.[1] The molecular mechanism of platelets in inflammatory responses mainly includes platelet activation, aggregation, and secretion of preexisting pro-inflammatory mediators. The activation of platelets mainly occurs through the interaction of the extracellular matrix, glycoprotein receptors (GPVI), and GPIb-IX-V on the platelet surface with von Willebrand factor (VMF) released by vascular endothelial cells, causing platelets to attach to collagen to form activated platelets. Subsequently, through the mediation of integrin αIIbβ3, fibrinogen binds to different αIIbβ3 at both ends through a “bridging” method (Fig. 1), ultimately leading to platelet aggregation.[2]
 Figure 1.:
Figure 1.: (P4) NET dependency mechanism of thrombosis in vivo (in OA_Figure 1).
Numerous studies have revealed the association between inflammation and haemostasis.[3] Platelet function is a source of granules in platelets. These granules are released by platelets, ultimately causing thrombus formation. Bactericidal protein in platelet granules released by platelets could be blind to bacteria, causing the elimination of these bacteria.[4] Past studies have mainly focused on the role of platelets in thrombosis, while relatively few studies have been conducted on their functions and regulatory mechanisms in the inflammatory center.
Although they showed that the primary function of platelets is haemostasis, the number and morphology of platelets are altered in sterile inflammation, especially autoinflammatory diseases.[5] It is unclear that platelets played a core role in different types of inflammation. This article aims to explore the role of platelets in the inflammatory center and its potential application value through a review of relevant literature. The study based on platelet granules described infectious and sterile inflammation separately, which indicated platelets had central regulation in different types of inflammation.
2. Material and methods 2.1. Research background and significance of plateletsPlatelets are a type of cellular fragments in blood, and they play an essential role in the physiological and pathological processes of the human body. In recent years, more and more studies have found that platelets play a vital role in inflammatory responses and can promote the occurrence and development of inflammation. This review aims to explore the role of platelets in the center of inflammation and provide us with new research directions and treatment strategies.
2.2. Research methodsThis article mainly conducts an in-depth study on the mechanism of platelets in inflammatory response through a literature review. By collecting and analyzing many relevant studies, we have revealed the mechanism of platelets’ role in inflammatory responses and how their interactions with various cell types contribute to the pathogenesis of vascular inflammation. Meanwhile, the study examined the current clinical anti-inflammatory treatment strategies to provide new ideas for clinical treatment.
3. Results 3.1. Platelets granulesThere are bluish-purple platelet granules in the central platelet, termed granulomere. A light blue heterogeneous zone in the surrounding platelet is called a hyalomere. There are 3 granule classes in granulomere: α-granules, β-granules, and lysosomes.[6]
3.2. α Granulesα granules included plasma protein, adhesion molecules, complement and complement activation regulators, complement binding proteins, hemostatic factors, angiogenic factors, anti-angiogenesis factors, growth factors, protease, necrosis factor, cell factors, cationic protein sterilization, etc. Plasma protein included coat protein such as clathrin, adaptor protein 1 (AP1), adaptor protein 2 (AP2), and protein required for vesicular transport such as the Neurexin/N-Ethylmaleimide-sensitive Factor (NSF), attachment protein receptor regulator, Sec1/Munc18 protein, and GTPase such as Rabs,[6] and IgG etc.[7] Adhesion molecules such as P-selectin, platelet endothelial cell adhesion molecule 1 (PECAM-1), and CD9-integrin alphaIIbbeta3 (GPIIb-IIIa) could mediate innate immunity.
Cationic bactericidal protein-mediated host immunity defence, including platelet factor 4 (CXCL4), thymosin-β4, CXCL7 derivatives, CCL5, thrombospondins 1, thrombospondins 2-Regulators of complement activation, complement, and complement binding protein could enhance immune cell function, including C3, C3b and C1-inhibitors.[8] The cytokine modulated platelets-leukocytes interaction, including CXCL1 (GRO-α), CXCL4, CXCL5 (ENA-78), CXCL7 (PBP, β-TG, CTAP-III, NAP-2), CXCL8 (IL-8), CXCL12 (SDF-1α), CCL2 (MCP-1), CCL3 (MIP-1α), and CCL5 (RANTES).[8]
Others need help with the classification of substances. For instance, hemostatic, angiogenesis, anti-angiogenesis, growth, proteases, and Necrosis factors included V, von Willebrand factor (vWF), and fibrinogen. Angiogenesis factors included angiogenin and VEGF. Anti-angiogenesis factors included vascular statin PF4. Growth factors included PDGF, bFGF, SDF1α. Protease included MMP2 and MMP9. Necrosis factors included TNFα, TNFβ.
3.3. Dense granulesDense granules included non-protein molecules such as ADP, 5-TH,[9] serotonin, polyphosphate,[10] pyrophosphate, calcium ions, melanosomes, granule fusion,[6] CD63 (LAMP 3) and LAMP 2, magnesium ions,[7] which played an essential role in expanding platelets.
3.4. Lysosomallysosomal included glycosidase and protein sterilization.[11] Peroxisomes included 1 to 3 lysosomes, such as beta aminocaproic glycosidase.[6] There are lysosomal platelets with cathepsin D, acid hydrolase, E100, and other lysosomal proteins.[7]
3.5. The role of platelets and leukocytes 3.5.1. The formation of immune thrombosis: neutrophils and plateletsThe interaction of platelets and neutrophils, besides neutrophil death, promoted the formation of immune thrombosis via neutrophil cell death, which could promote innate immune responses.[12] Neutrophil extracellular traps (NETs), DNA, histone, and particulate components are favorable factors promoting the formation of immune thrombosis, which is considered overlapping mechanisms between the immune system signals and the coagulation signals.[13–15] The risk of thrombosis increases when the innate immune induced by immune-inflammatory thrombosis can hardly take control of severe infection.[9]
Stuart Wallis showed that efb68-87 could blind directly to P-selectin and inhibit interactions of platelets with leukocytes that could lead to PLA and NET formation.[16] Huilian Chen showed that fruit flow could inhibit platelet function by suppressing Akt/GSK3β, Syk/PLCγ2 and p38 MAPK phosphorylation in collagen-stimulated platelets.[17] Min Li reanalyzed the GSE45111 dataset of a spectrum of asthma, and they showed that platelet activation could enhance the Th2 immune response and induce lung inflammation through δ, α, λ granule released by platelets.[18] Maximilian Mauler used two murine models of acute inflammation, and they showed that LPS challenge in Tph1-/- mice prevented leukocyte recruitment and reduced platelet neutrophil complex formation.[9] David M. Chesko and Wilgus found through 10 years of a retrospective study that platelet depletion enhanced the recruitment of T cells and macrophages. In the absence of platelets, mediators produced by other cells could overcome the lack of platelet-derived mediators in thrombocytopenic mice.[19] Dimitra Gialamprinou thought that prostaglandin E1 synthesis modulated by platelets affected hypersensitive response.[20]
3.6. Platelets mediated sterile inflammationCell injury probably induced sterile inflammation. Sterile inflammation had similar pathological progress. Atherosclerosis is a joint sterile inflammation.[5] Autoinflammatory Disease also causes sterile inflammation, such as chronic tophaceous gout. Cell injury activated Damage-Associated Molecular Patterns via monocyte/macrophage, leading to the inflammatory response. Cell injury released some substances such as IL-1α, S100 protein, heat shock protein, and dsDNA, which could activate some signal pathways; for example, MAP Kinase activation induced the role of NF-κB.[21] Meanwhile, cell injury through dendritic cell activation released soluble platelet factors influencing inflammatory responses.[22] However, active platelets could induce the formation of neutrophil extracellular traps, thus activating inflammatory response.[23] Eithne Nic and Riogh suggested that an appropriate platelet function assay could guide future therapy for patients with inflammatory arthritis.[24] Serena Bianchi proved that platelets play a vital role in hemostasis, immune regulation, and repair mechanisms.[25] H. ATLI1 et al found that neutrophil-to-lymphocyte ratio and platelet-to-lymphocyte ratio are related to the onset of proliferative diabetic retinopathy, and neutrophil-to-lymphocyte ratio could predict microvascular inflammation.[26] Recep Yilmaz found that neutrophil/lymphocyte ratio but not platelet/lymphocyte ratio and mean platelet volume can indicate subclinical inflammation in patients with Familial Mediterranean Fever.[27] Abderrahim Nemmar found via studying platelet activation by the neutrophil-released proteases cathepsin G and elastase that it potently activated platelets via an ADP-mediated mechanism, which potently enhanced a condition accompanied by local and systemic inflammation.[28]
3.7. Platelets mediated inflammation of the pathogenStudies have found that platelets are both “inciters” and “participants” in inflammatory responses. In the inflammatory reaction, platelets are activated by invading pathogens or inflammatory factors. The activated platelets are in the bloodstream at sites infected by microorganisms. Platelet aggregation can be observed in inflammatory exudates and inflammatory tissues. They share common antigens, regulating inflammatory damage and affecting leukocytes’ biological behavior and inflammatory signaling pathways to varying degrees.[29] In addition, platelets are the earliest and most numerous cells recruited to the subendothelial blood vessels in the inflammatory response.[30] They interact with neutrophils, monocytes, antigens, antigen-related products, and inflammatory vascular endothelium, leading to microvascular occlusion and thrombosis, spread of inflammation and damage to blood vessel walls.[30]
3.7.1. The role of platelets with bacteriaThe interaction of platelets with bacteria-activated platelets and formatted immune thrombosis to prevent a spreading bacterium and, meanwhile, pro-inflammatory mediator released by platelets could play a significant role in bacterial infections immunity.[31] For instance, streptococcus purulent is first snared by fibrinogen and then combined with GPIIb/IIIa signals from platelets, thus activating platelets.[4] When platelets act, platelet volume expansion induces the aggregation of platelets, leading to the capture of bacteria. Next, releasing inflammatory mediators in platelet granulomere could activate leukocytes to accomplish bacterial infection immunity. These inflammatory mediators included regulated upon activation of normal T cell expressed and secreted factor (RANTES), PF4, soluble CD40-ligand (sCD40L), soluble P-selectin, PDGF and ADP.[32,33]
3.7.2. The role of platelets with fungusPlatelets could be blind to a fungus-associated substance that significantly affects fungus infection immunity.[34,35] For example, protease-activated receptors (PARs) and toll-like receptors (TLRs) could identify β-1,3 glucan or β-galactomannan leading signaling cascade.[36] Meanwhile, β-glucan and α-glucan could protect the human body against Aspergillus infection.[37] Bactericidal substances released by platelets, such as 5-HT, are considered antibacterial peptides that exert immune function.[38]
3.7.3. The role of platelets with virusPlatelets mediate virus infection immunity by regulating the Toll-like receptor pathway.[10] Platelets activated Toll-like receptors through the phagocytosis of the virus. Thrombocytopenia after severe viral infections manifests antiviral immunity function decline.[39] The integrin receptor probably affected the platelet phagocytosis response significantly.[40] Participating in human immunodeficiency virus research revealed that platelets could be controlled in a slow decline of viral load, and virus particles are found in platelets’ cytoplasm, which confirmed platelets’ phagocytosis response.[41] Sokratis A. Apostolidis blocking the signaling of the FcgRIIa-Syk and C5a-C5aR pathways on platelets revealed a crucial role in platelet-mediated immune thrombosis.[42] Li Tianyang showed that platelets could mediate inflammatory monocyte activation by SARS-CoV-2 spike protein.[43] Ana Kasirer-Friede showed that platelet SHARPIN could regulate platelet adhesion and inflammatory responses through associations with αIIbβ3 and LUBAC and modulate CD4+ T-cell function in COVID-19 through a PD-L1-dependent mechanism.[44,45] To further investigate platelet reactivity in COVID-19, Alexey A. Martyanov analyzed 46 COVID-19 patients’ data and showed that coagulation dysfunction of COVID-19 is associated with intravascular coagulation-induced refractoriness.[46]
3.7.4. The role of platelets with parasiteParticipating in malaria research revealed that platelets via the release of platelet plasmodial factor, platelet factor 4 (PF4) and the red cell-expressed Duffy-antigen molecule could mediate parasite immunology.[47,48]
First, platelets are blinded with infection erythrocytes and induce the interaction of plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) and scavenger receptor CD36. Then, this infection protein marker is transferred to the erythrocyte surface, leading to identification and elimination.[49,50]
3.7.5. Platelets and complementConsumption of little complement-related substance during the uptake of plasma by platelets could induce platelets associated with the complement pathway. The formation of C3b, C3a, and C5b-9 activated the complement pathway, which is induced by platelet activation and P-Selectin.[51] Meanwhile, chondroitin sulfate released by platelets could activate the complement pathway.[52] However, gC1qR expressed by platelets could also activate the classical complement one.[53] The interaction of platelets with complement extensively promoted pathogen immune killing by platelets.
3.8. The role of platelets in tumourAdhesive proteins modulated platelet-tumor interaction. For example, active platelets are covered on the tumor cell’s surface. This formation of platelets-tumor micro-thrombocytosis could be conducive to protecting tumor cells against the mechanical shear of the blood’s circulation.[54] Meanwhile, by dynein, tumor cells uptake platelets CD42a, thus obtaining all metabolic substances by phagocytosis platelets in platelets’ cytoplasm.[55] Podoplanin is a sialylated membrane glycoprotein. It could bind to C-type lectin-like receptor two on platelets, which mediated platelet activation. Podoplanin expression on cancer cells is involved in metastasis; the low molecular weight protein tyrosine phosphatase (LMWPTP) at the cell level affects tumor-platelet interaction.[56] Upregulating LMWPTP at the cell level could promote tumor cell proliferation.
Interestingly, tumor cell proliferation upregulated LMWPTP at the cell level via positive feedback, which enhanced tumor invasion and chemotherapy resistance. In addition, protein tyrosine kinases could be a potential therapeutic target.[57] Xiaowei Liu showed that platelet protects angiotensin II-driven abdominal aortic aneurysm formation by inhibiting inflammation.[58] Dina Ali Hamad combined blood indexes of systemic inflammation as a mirror to admission to the intensive care unit in COVID-19 patients. She found that SIRI is associated with clinical results, which predicted survival rates of breast cancer and gastric cancer.[59] SIRI is defined in the following formula: SIRI = N × P/L, where N could represent neutrophils, P-platelets, and L-lymphocytes. Interestingly, this effective parameter fully reflected the balance between the host’s immune and inflammatory status.[59]
3.9. Platelets induced epilepsy immuneThe presence of the lymphatics in the brain received very little attention. Because of the tight junction and non-fenestrated capillaries, the blood-brain barrier (BBB) could prevent the passage of most substances of the blood, especially immune cells, which construct an immuno-isolation membrane. Thus, lymphatics in the brain are essential structures in the brain region.[60] When epilepsy occurs, the local electrical activity of brain cells is abnormal. Dysregulation of brain-electric activity homeostasis would harm the brain tissue, such as cell membrane damage and cell damage. When brain tissue is damaged, especially BBB, those substances absent from brain tissue are released by BBB. At that moment, those substances could activate platelets to BBB repair, thus via TLR, limiting the implications to movement, which could abstract innate immune cells to eliminate those substances. There are usually astrocyte cells, microglial cells, and mast cells in this remarkable immune response.
Interestingly, those immune cells could release a lot of S100b.[61] That S100 b also, via the super pathway, activates neuroimmune response. For example, TRAAF6 started TLR7/8 or 9 to induce NFκB and MAP kinase, leading to amplified immune responses.[62] A continuous immune response could cause inflammatory immune infiltration, which damages brain tissue again, especially endothelial cells of BBB, and the extent of the damage is variable. Injured BBB reduced the protective effect, which caused out-brain substances to enter the brain area, leading to chronic immune injury. Surprisingly, gamma globulin levels remained high in the blood, which could alleviate this destruction of BBB.[61] If BBB is repaired by gamma globulin currently, how is the immune injury reaction in the brain area? We predicted that compared with the out-brain immune system, the local immune response of brain area lymphatics could have a higher dominance rank, which caused the local immune response of brain area lymphatics to eliminate out-brain substances. Moreover, the brain area’s immune homeostasis is restored.
4. DiscussionThe literature review proves that platelets are critical in orchestrating inflammatory responses. The ability of platelets to act as central hubs in inflammation is mediated through their extensive repertoire of surface receptors, which facilitate dynamic interactions with various immune cells. These interactions are not merely passive but involve active recruitment and modulation of leukocytes, contributing to amplifying or attenuating the inflammatory cascade.
Platelets’ release of soluble mediators, such as cytokines and chemokines, further underscores their active participation in inflammation. These mediators have diverse effects on the immune system, influencing cell migration, differentiation, and function. The involvement of platelets in the formation of NETs represents another dimension of their role in inflammation, linking them to both pathogen clearance and the potential for tissue damage.
4.1. The role of platelets in the center of inflammationThe role of platelets in the inflammatory center is mainly reflected in the following aspects:
Platelets promote the recruitment and activation of inflammatory cells (such as monocytes, neutrophils, etc.) by releasing various biologically active substances, such as growth factors, chemokines, and prostaglandins. Platelets interact with vascular endothelial cells, increasing vascular permeability and making it easier for inflammatory cells and inflammatory factors to enter tissues, thus aggravating the inflammatory response. Platelets regulate the duration and intensity of inflammatory responses by affecting the phenotype and function of immune cells. 4.2. The potential value of platelets in the treatment of inflammatory diseasesClinical implications of platelet involvement in inflammation are profound, with numerous studies linking dysregulated platelet activity to the pathogenesis of inflammatory diseases. This phenomenon suggests that platelets could be viable targets for therapeutic intervention. However, the complex platelet functions in inflammation challenge the development of such therapies. Inhibition of platelet activity could potentially ameliorate inflammatory disease symptoms, but it also risks impairing haemostasis and host defence mechanisms.
4.3. Research prospectsThis review reveals a new role of platelets in the center of inflammation and provides a theoretical basis for further research on the part of platelets in inflammatory diseases. However, the specific molecular mechanisms and regulatory networks of platelets in inflammatory responses still require in-depth study. In addition, the clinical application of platelet-targeted therapy strategies in inflammatory diseases also deserves attention. Future research should dissect the specific signaling pathways and molecular interactions that govern platelet function in inflammation. This situation could lead to identifying novel therapeutic targets that modulate platelet activity without compromising their essential roles in haemostasis and immunity. Additionally, understanding the temporal dynamics of platelet involvement in inflammation could provide insights into the transition from acute to chronic inflammatory states, offering opportunities for stage-specific therapeutic interventions.
5. ConclusionPlatelets played a core role in developing an immunologic inflammatory response. Formation of immune thrombosis by capturing targets activated platelets causes the release of signals to accomplish immune clearance. On one hand, platelets could release complement and bactericidal proteins, thus inducing pathogens’ death. On the other hand, they could initiate the complement pathway.
Moreover, platelets via DAMPS activated immunological inflammation. Meanwhile, platelets via S100b could induce epilepsy immunity. All in all, platelets are widely attended to in all kinds of immunological inflammation. Furthermore, platelets could reflect the exemption. Therefore, platelets could keep a range of exceptional levels beneficial for stabilizing immunity function.
We look forward to future research to achieve more breakthroughs in this field and bring new hope for treating inflammatory diseases.
AcknowledgmentsThe authors thank Prof Fei Zhao of Northwest Minzu University, who helped them with this article.
Author contributionsConceptualization: Yan Bo.
Data curation: Yan Bo.
Investigation: Yan Bo.
Methodology: Beilei Li.
Project administration: Ren Sha.
Resources: Ren Sha.
Validation: Yan Bo, Haodong Yu.
Visualization: Yan Bo, Haodong Yu.
Writing – original draft: Yan Bo.
Writing – review & editing: Qingyang Lu, Chuhan Miao.
References [1]. Hunter P. The inflammation theory of disease. The growing realization that chronic inflammation is crucial in many diseases opens new avenues for treatment. EMBO Rep. 2012;13:968–70. [2]. Shanley DK, Kiely PA, Golla K, et al. Pregnancy-specific glycoproteins bind integrin αIIbβ3 and inhibit the platelet-fibrinogen interaction. PLoS One. 2013;8:e57491. [3]. Kapoor S, Opneja A, Nayak L. The role of neutrophils in thrombosis. Thromb Res. 2018;170:87–96. [4]. Shannon O, Hertzén E, Norrby-Teglund A, et al. Severe streptococcal infection is associated with M protein-induced platelet activation and thrombus formation. Mol Microbiol. 2007;65:1147–57. [5]. Rock KL, Latz E, Ontiveros F, et al. The sterile inflammatory response. Annu Rev Immunol. 2010;28:321–42. [6]. Sharda A, Flaumenhaft R. The life cycle of platelet granules [version 1; peer review: 2 approved]. F1000 Res. 2018;7:236-. [7]. King SM, Reed GL. Development of platelet secretory granules. Semin Cell Dev Biol. 2002;13:293–302. [8]. Blair P, Flaumenhaft R. Platelet α-granules: basic biology and clinical correlates. Blood Rev. 2009;23:177–89. [9]. Mauler M, Schanze N, Krauel K, et al. Peripheral serotonin lacks effects on endothelial adhesion molecule expression in acute inflammation. J Thromb Haemost. 2022;20:222–9. [10]. Banerjee M, Huang Y, Joshi S, et al. Platelets endocytose viral particles and are activated via TLR (Toll-Like Receptor) signaling. Arterioscler Thromb Vasc Biol. 2020;40:1635–50. [11]. Rendu F, Brohard-Bohn B. The platelet release reaction: granules’ constituents, secretion and functions. Platelets. 2001;12:261–73. [12]. Kerlin BA, Yan SB, Isermann BH, et al. Survival advantage associated with heterozygous factor V Leiden mutation in patients with severe sepsis and in mouse endotoxemia. Blood. 2003;102:3085–92. [13]. Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107:15880–5. [14]. Kambas K, Mitroulis I, Ritis K. The emerging role of neutrophils in thrombosis-the journey of TF through NETs. Front Immunol. 2012;3:385. [15]. Reges K, Engelmann B, Bidzhekov K, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16:887–96. [16]. Wallis S, Wolska N, Englert H, et al. A peptide from the staphylococcal protein Efb binds P-selectin and inhibits the interaction of platelets with leukocytes. J Thromb Haemost. 2022;20:729–41. [17]. Chen H, Zhang S, Wang H, et al. Fruitflow inhibits platelet function by suppressing Akt/GSK3β, Syk/PLCγ2 and p38 MAPK phosphorylation in collagen-stimulated platelets. BMC Complement Med Ther. 2022;22:75. [18]. Li M, Zhu W, Saeed U, et al. Identification of the molecular subgroups in asthma by gene expression profiles: airway inflammation implications. BMC Pulm Med. 2022;22:29. [19]. Chesko DM, Wilgus TA. Immune cells in cutaneous wound healing: a review of functional data from animal models. Int J Mol Sci. 2022;23:2444. [20]. Gialamprinou D, Mitsiakos G, Katsaras GN, et al. Neonatal sepsis and hemostasis. Diagnostics (Basel). 2022;12:261. [21]. Orlova VV, Choi EY, Xie C, et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007;26:1129–39. [22]. Hilf N, Singh-Jasuja H, Schwarzmaier P, et al. Human platelets express heat shock protein receptors and regulate dendritic cell maturation. Blood. 2002;99:3676–82. [23]. Maugeri N, Campana L, Gavina M, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12:2074–88. [24]. Riogh EN, Dunne E, Cowley S, et al. Dynamic platelet function: a novel biomarker in inflammatory arthritis? PLoS One. 2022;17:e02618–25. [25]. Bianchi S, Torge D, Rinaldi F, et al. Platelets’ role in dentistry: from oral pathology to regenerative potential. Biomedicines. 2022;10:218. [26]. Atli H, Onalan E, Yakar B, et al. Predictive value of inflammatory and hematological data in diabetic and non-diabetic retinopathy. Eur Rev Med Pharmacol Sci. 2022;26:76–83. [27]. Dinçer ABK, Gülöksüz EGA, Sezer S, et al. Neutrophil/lymphocyte ratio but not platelet/lymphocyte ratio and mean platelet volume can be an indicator of subclinical inflammation in patients with Familial Mediterranean Fever. Egypt Rheumatologist. 2022;44:215–8. [28]. Nemmar A, Hoylaerts MF. Neutrophil cathepsin G enhances thrombogenicity of mildly injured arteries via ADP-mediated platelet sensitization. Int J Mol Sci. 2022;23:744. [29]. Zhang H, Tang W, Wang S, et al. Tetramethylpyrazine inhibits platelet adhesion and inflammatory response in vascular endothelial cells by inhibiting P38 MAPK and NF-κB signaling pathways. Inflammation. 2020;43:286–97. [30]. Kriplani A, Pandit S, Chawla A, et al. Neutrophil–lymphocyte ratio (NLR), platelet–lymphocyte ratio (PLR) and lymphocyte–monocyte ratio (LMR) in predicting systemic inflammatory response syndrome (SIRS) and sepsis after percutaneous nephrolithotomy (PNL). Urolithiasis. 2022;50:341–8. [31]. Iba T, Levy JH. Inflammation and thrombosis: roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J Thromb Haemost. 2018;16:231–41. [32]. McNicol A, Agpalza A, Jackson ECG, et al. Streptococcus sanguinis-induced cytokine release from platelets. J Thromb Haemost. 2011;9:2038–49. [33]. Herzberg MC, Krishnan LK, MacFarlane GD. Involvement of α2-adrenoreceptors and G proteins in the modulation of platelet secretion in response to Streptococcus sanguis. Crit Rev Oral Biol Med. 1993;4:435–42. [34]. Sorgo AG, Heilmann CJ, Brul S, et al. Beyond the wall: Candida albicans secret(e)s to survive. FEMS Microbiol Lett. 2013;338:10–7. [35]. Girard V, Dieryckx C, Job C, et al. Secretomes: the fungal strike force. Proteomics. 2013;13:597–608. [36]. Roeder A, Kirschning CJ, Rupec RA, et al. Toll-like receptors as key mediators in innate antifungal immunity. Med Mycol. 2004;42:485–98. [37]. Fontaine T, Delangle A, Simenel C, et al. Galactosaminogalactan, a new immunosuppressive polysaccharide of Aspergillus fumigatus. PLoS Pathog. 2011;7:e1002372–72-e. [38]. Perkhofer S, Niederegger H, Blum G, et al. Interaction of 5-hydroxytryptamine (serotonin) against Aspergillus spp. in vitro. Int J Antimicrob Agents. 2007;29:424–9. [39]. Health and Medicine – Hematology; Researchers at University Medical Center Utrecht Report Findings in Hematology (Influenza-induced thrombocytopenia is dependent on the subtype and sialoglycan receptor and increases with virus pathogenicity). Virus Weekly. 2020:1632. [40]. Shimony N, Elkin G, Kolodkin-Gal D, et al. Analysis of adenoviral attachment to human platelets. Virol J. 2009;6:25. [41]. Real F, Capron C, Sennepin A, et al. Platelets from HIV-infected individuals on antiretroviral drug therapy with poor CD4 T cell recovery can harbor replication-competent HIV despite viral suppression. Sci Transl Med. 2020;12. [42]. Apostolidis SA, Sarkar A, Giannini HM, et al. Signaling through FcγRIIA and the C5a-C5aR pathway mediates platelet hyperactivation in COVID-19. bioRxiv. 2021. [43]. Li T, Yang Y, Li Y, et al. Platelets mediate inflammatory monocyte activation by SARS-CoV-2 spike protein. J Clin Invest. 2022;132:1–10. [44]. Kasirer-Friede A, Peuhu E, Ivaska J, et al. Platelet SHARPIN regulates platelet adhesion and inflammatory responses through associations with αIIbβ3 and LUBAC. Blood Adv. 2022;6:2595–607. [45]. Paletta A, Di Diego García F, Varese A, et al. Platelets modulate CD4+ T-cell function in COVID-19 through a PD-L1 dependent mechanism. Br J Haematol. 2022;197:283–92. [46]. Martyanov AA, Boldova AE, Stepanyan MG, et al. Longitudinal multiparametric characterization of platelet dysfunction in COVID-19: effects of disease severity, anticoagulation therapy and inflammatory status. Thromb Res. 2022;211:27–37. [47]. McMorran BJ, Wieczorski L, Drysdale KE, et al. Platelet factor 4 and Duffy antigen required for platelet killing of plasmodium falciparum. Science. 2012;338:1348–51. [48]. Chaudhuri A, Zbrzezna V, Polyakova J, et al. Expression of the Duffy antigen in K562 cells. Evidence that it is the human erythrocyte chemokine receptor. J Biol Chem. 1994;269:7835–8. [49]. McMorran BJ, Burgio G, Foote SJ. New insights into the protective power of platelets in malaria infection. Commun Integr Biol. 2013;6:e23653. [50]. Peyron F, Polack B, Lamotte D, et al. Plasmodium falciparum growth inhibition by human platelets in vitro. Parasitology. 1989;99 Pt 3:317–22. [51]. Del Conde I, Crúz MA, Zhang H, et al. Platelet activation leads to activation and propagation of the complement system. J Exp Med. 2005;201:871–9. [52]. Hamad OA, Nilsson Ekdahl K, Nilsson PH, et al. Complement activation triggered by chondroitin sulfate reelased by thrombin receptor-activated platelets. J Thromb Haemost. 2008;6:1413. [53]. Yin W, Ghebrehiwet B, Peerschke EIB. Expression of complement components and inhibitors on platelet microparticles. Platelets. 2008;19:225–33. [54]. Palacios-Acedo A-L, Langiu M, Crescence L, et al. Platelet and cancer-cell interactions modulate cancer-associated thrombosis risk in different cancer types. Cancers. 2022;14:730. [55]. Martins Castanheira N, Spanhofer AK, Wiener S, et al. Uptake of platelets by cancer cells and recycling of the platelet protein CD42a. J Thromb Haemost. 2022;20:170–81. [56]. Sasano T, Gonzalez-Delgado R, Muñoz NM, et al. Podoplanin promotes tumor growth, platelet aggregation, and venous thrombosis in murine models of ovarian cancer. J Thromb Haemost. 2022;20:104–14. [57]. Bhullar JS. Gas
留言 (0)