Study design and patients
This was a 3-month, multicenter, prospective, open-label, non-interventional pharmacoepidemiologic study conducted at four centers in Barcelona, Spain, from November 5, 2020, to February 23, 2023. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Fundació Unio Catalana Hospitals local ethics committee approved the study (code CEI 19/33), and it was accepted by all the sites. All patients provided written informed consent before study participation.
Eligible participants were those who attended pain units on an outpatient basis, showed an ineffective response to the long-lasting medication for depression they were receiving at that time, and/or had considerable adverse effects to the antidepressants. The inclusion criteria were male or female participants, aged ≥ 18 years, already diagnosed with MDD, with a 9-item Patient Health Questionnaire (PHQ-9) score ≥ 15, with an associated CP condition (visual analogic scale [VAS] score ≥ 4), and having concomitant medication to treat habitual pain, with no changes during the study. This analgesic medication consists of minor opioids, nonsteroidal anti-inflammatory drugs (NSAIDs), paracetamol, and/or metamizole (any of them inducing undesirable effects such as drowsiness, dry mouth, constipation, and disorientation). Patients were required to read and understand the Patient Information sheet and sign an Informed Consent form. Patients with any contraindications specified in the Vortioxetine SPC, cognitive impairment that mades it impossible for them to sign the informed consent form, or suffering from significant acute or unstable disease or unstable medical condition that could compromise their participation in the study according to the investigator's judgment were ineligible for study participation. Patients who were members of the staff involved in the study or their immediate family, or a subordinate (or an immediate family member of a subordinate) of the staff involved in the study, who were previously enrolled in this study, or who participated in another ongoing clinical trial were also excluded. In addition, patients who had been receiving treatment with major opioids or, in the opinion of the investigator, were unable to comply with the study protocol were not considered.
To reduce the risk of selection bias, the following measures were implemented: (1) patients were consecutively included in the study; (2) investigators were trained to respect the protocol and remain neutral during the evaluation of the patients to be included in the study; (3) several centers were included to reflect the diverse range of practices, which was expected to reduce bias linked to systematic errors; (4) a very specific sample was chosen with little variability in baseline characteristics.
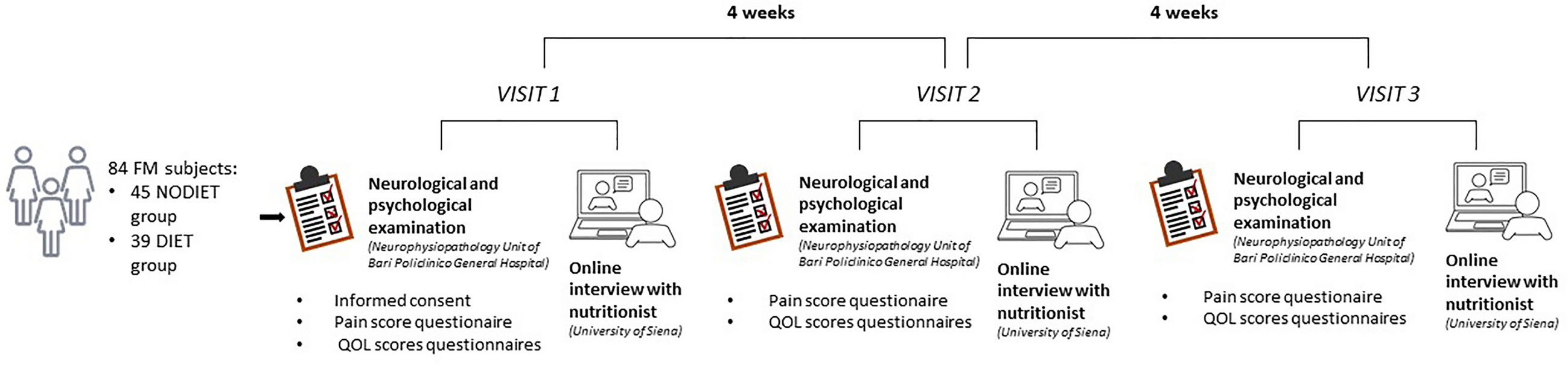
Vortioxetine baseline treatment was 10 mg/day. Fifteen days after the first dose, patients received a phone call to evaluate their clinical evolution, and the dose could be increased to the maximum therapeutic dose (20 mg/day) or decreased to 5 mg/day based on individual patient response at the investigator’s judgment. All patients continued with pain medication during the study, as per common clinical practice. Study assessments and follow-ups were performed at three visits: at baseline, 1 month (± 2 weeks), and 3 months (± 2 weeks) of vortioxetine treatment (visits 1, 2, and 3, respectively).
Study Assessments
The primary objective was to assess the improvement in depressive symptoms using the 9-item Patient Health Questionnaire (PHQ-9). PHQ-9 consists of nine items with Likert-type responses ranging from 0 to 3, relative to the last two weeks. A total score between 0 and 27 is obtained:1–4, 5–9, 10–14, 15–19, and 20–27 represent minimal, mild, moderate, moderately severe, and severe depression, respectively [45].
Secondary objectives were to assess improvement in pain and treatment satisfaction in the patients. To this aim we used the following tests:
Visual analogic scale (VAS) [46] helps determine the intensity of pain to be measured with maximum reproducibility between observers. It consists of a 10-cm horizontal line, the ends of which are the extreme expressions of a symptom. The left end shows the absence of pain or lowest intensity, and the right end shows the highest intensity. The patient is asked to mark the point on the line indicating the intensity, which is measured with a millimeter ruler in centimeters or millimeters. Thus, a total measure between 0 and 10 is obtained: 0–3, 4–7, and 8–10 represent mild, moderate, and severe pain, respectively. Patients with a VAS score < 4 suffer from mild pain, which thus should not lead to major depression. Consequently, they were excluded from this study.
Brief Pain Inventory (BPI) [47] consists of 10 items. It is a multidimensional pain assessment instrument that provides information on pain intensity and its interference with activities of daily living. It also assesses the description, location, and level of pain relief associated with the treatment.
The Chronic Pain Coping Inventory (in Spanish, “Cuestionario de Afrontamiento del Dolor” [CAD]) [48] is a questionnaire on strategies to cope with pain, built using an entirely Spanish sample of patients with CP. It consists of 31 items grouped into six dimensions: self-affirmation, readiness to find information, religion, distraction, catharsis, and mental self-control. Religion and catharsis dimensions are grouped into “passive” pain coping and the remaining dimensions into “active” pain coping. The items are assessed on a 5-point agree/disagree scale.
Clinical Global Impression (CGI) Scale [49] was used to assess the overall disease severity and clinical improvement. In the current study, the Improvement (CGI-I) clinician-rated subscale was used. The Patient Global Impression Improvement (PGI-I) scale [50] consists of a single question asking the patient to rate the relief obtained with treatment. Both scales range from 1 (very much improved) to 7 (very much worse).
Satisfaction with Medicines Questionnaire (SATMED-Q) [51] is a 17-item questionnaire with six dimensions: treatment effectiveness, convenience of use, impact on daily activities, medical care, global satisfaction, and undesirable side effects. It measures the level of patient satisfaction with health care and treatment.
Adverse events (AEs) associated with vortioxetine were described during treatment. The summary included the dates of start and end, outcome, causality relationship, and severity criteria.
BPI scale and PHQ-9, CAD, and SATMED-Q questionnaires were administered at baseline and at 3 months and VAS, CGI, and PGI scales at baseline and at 1 and 3 months.
Statistical Analysis
The sample size was calculated based on the primary variable of the study: the change from baseline to month 3 in the PHQ-9 total score. With an SD of 5, the objective was to recruit 100 patients to obtain a 95% confidence interval (95% CI) with an accuracy error of ≤ 1.
The evaluable population comprised all eligible patients who had taken at least one dose of vortioxetine and had at least one post-baseline datum on the primary endpoints, including those patients with protocol violations or who discontinued the study. The safety population included all patients who received at least one dose of the study treatment.
Descriptive statistical analyses were performed for all variables. Continuous variables were described by the number of valid cases, mean, standard deviation (SD), median, 25th and 75th percentiles (P25-P75), and minimum and maximum. On the other hand, categorical variables were described by absolute and relative frequencies of each category over the total number of valid values (N). In case of missing values, the number per group was described. Comparisons of categorical variables were made using ANOVA, chi-square test, or Fisher's exact test, as applicable. For continuous variables, the Student t-test for paired data or the Mann-Whitney U-test was used depending on the type of data. A bilateral statistical significance level of 0.05 was used for all comparisons.
Statistical analysis was performed following the principles specified in the ICH E9 guidelines as well as in the standards of good clinical practice. Statistical analysis was performed using SAS (Statistical Analysis System), version 9.4, on a Windows platform.





留言 (0)