
記住我






We performed a prospective study on patients diagnosed with pancreatic neoplasms at Kyushu University Hospital between April 2020 and March 2023. Informed consent was obtained from all patients. In this study, we focused on PDAC. Clinical data of patients with PDAC, including information about age, sex, pathological diagnosis, presence of metastases, Union for International Cancer Control (UICC) staging, treatment procedures, and prognosis, were obtained from electronic medical records. This study was approved by the Ethics Committee of Kyushu University (approval number: 22121-00) and was conducted according to the Ethical Guidelines for Human Genome/Gene Research enacted by the Japanese Government and the Helsinki Declaration.
SamplesTumor samples were obtained by endoscopic ultrasound-guided fine needle biopsy (EUS-FNB), liver biopsy, surgical resection, abdominal paracentesis, and endoscopic retrograde cholangiopancreatography (ERCP). Single-pass EUS-FNB was performed using a 19G or 22G needle (Acquire™; Boston Scientific, Marlborough, MA, USA; TopGain; MediGlobe, Tempe, AZ, USA) as previously reported [29]. Percutaneous biopsy of liver metastases was performed using a 16G or 21G lancet needle. The other samples obtained were as follows: 30–50 mm3 of surgical resection tissue; 100–200 mL of ascites via percutaneous abdominal paracentesis; and 5–10 mL of pancreatic juice via ERCP.
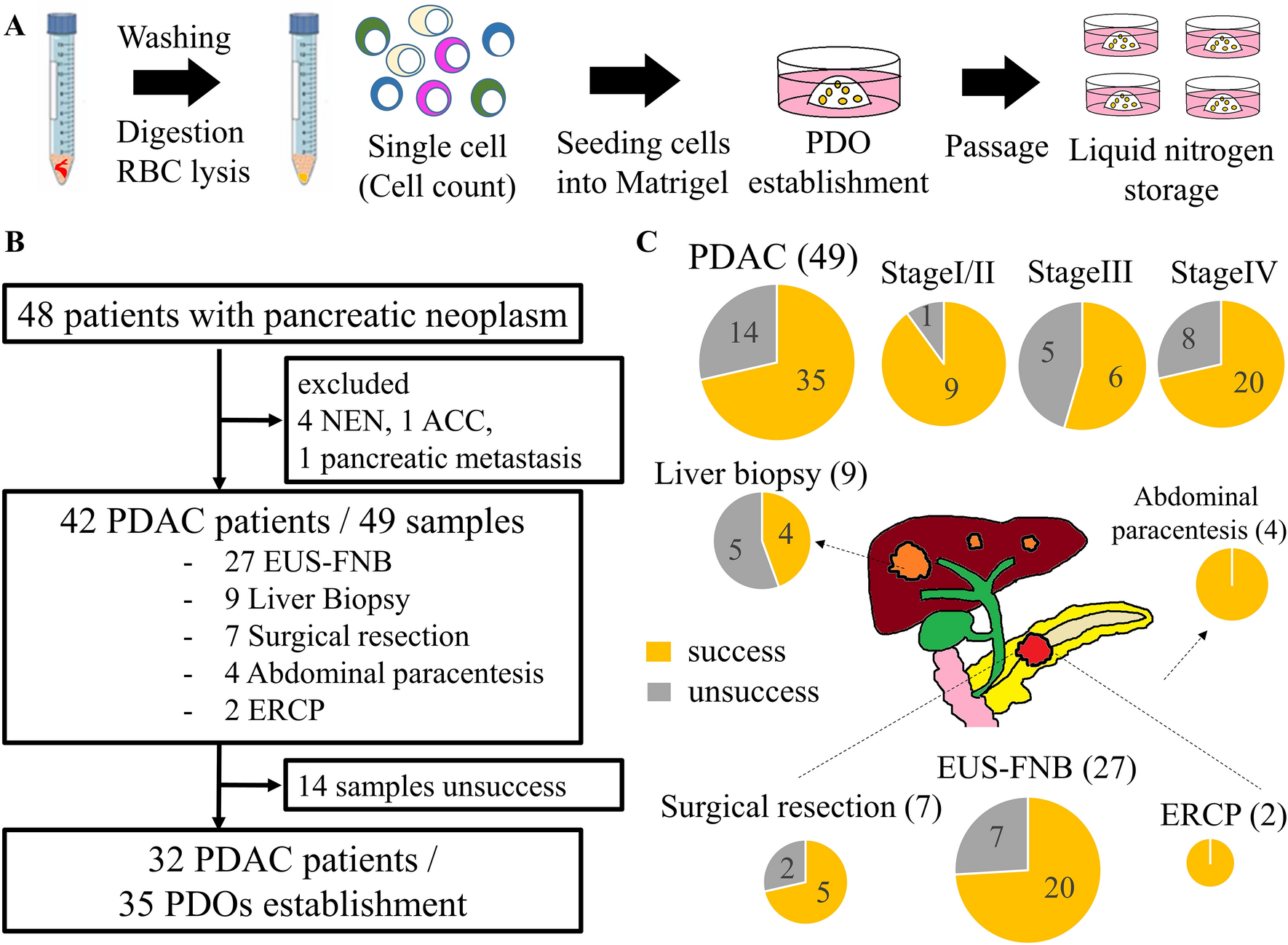
PDOsPDOs were established according to previous reports, with slight modifications [30, 31]. Solid PDAC samples obtained via EUS-FNB, surgical resection, and liver biopsy were washed with ice-cold phosphate-buffered saline (PBS). The surgically resected tissues were minced into approximately 1 mm3 fragments before digestion. Samples were digested into single cells using Liberase TH (20 min) and TrypLE Express (10 min) at 37 °C in a water bath (with pipetting every 5 min). After washing with basal medium (Supplementary Table S1), the red blood cells (RBCs) were hemolyzed in RBC lysis buffer for 10 min. Liquid samples, such as ascites and pancreatic juice obtained via ERCP, were washed with basal medium. The dissociated cells were embedded in Matrigel (356,231; Corning, NY, USA) and cultured in complete medium (Supplementary Table S1) in a 24-well plate (353,504; Corning) in a 37 °C incubator with 5% CO2 (Fig. 1A).
Fig. 1
A Illustration of the process involved in PDO establishment. Tumor samples underwent digestion into single cells, followed by embedding in Matrigel, and culture in complete medium to generate PDOs (patient-derived organoids). B Flowchart of this study. NEN neuroendocrine neoplasm, ACC acinar cell carcinoma, PDAC pancreatic ductal adenocarcinoma, EUS-FNB endoscopic ultrasound-guided fine-needle biopsy, ERCP endoscopic retrograde cholangiopancreatography (C), Success rates of PDO establishment using different sampling methods, highlighting the utility of EUS-FNB in PDO generation
The culture medium was changed every 3–4 d. For passaging, the PDOs were collected and dissociated by digestion with TrypLE Express for 3–5 min. The cells dissociated from the PDOs were replated with fresh Matrigel and cultured in a complete medium. PDOs were cultured without EGF after passaging to enrich the KRAS mutations, as reported previously [22]. Successful establishment of PDOs was defined as success in passage five times. Images of the PDOs were acquired via fluorescence microscopy (BZ-X710; Keyence, Osaka, Japan), and analyzed using the ImageJ software (National Institutes of Health, MD, USA). The PDOs were assigned in the order of their establishment.
Total RNA extraction and quantitative RT-PCR (qRT-PCR)Total RNA was extracted from the frozen samples of surgically resected tumors and the cultured PDOs using the ISOGEN reagent (NIPPON GENE, Tokyo, Japan) according to the manufacturer’s instructions. qRT-PCR was performed using the CFX Connect Real-Time System (Bio-Rad Laboratories, Hercules, CA, USA). The double-stranded DNA-specific dye SYBR Green I was incorporated into the PCR buffer provided in TB Green Premix EX Taq II (Takara Bio Inc., Shiga, Japan) to enable quantitative detection of the PCR product. Transcript levels were determined using the ΔΔCt method and normalized to that of ACTB mRNA. The primer sequences are listed in Supplementary Table S2.
RNA-seq and data analysisThe NEBNext rRNA Depletion Kit (New England Biolabs, Ipswich, MA, USA) was used to deplete ribosomal RNA from total RNA (500 ng), and the RNA was converted to an Illumina sequencing library using the NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs). The library was validated using a Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) to determine the size distribution and concentration, and sequenced on the NextSeq 500 (Illumina, San Diego, CA, USA) with the paired-end 36-base read option. Reads were mapped to the human reference genome hg19 using STAR version 2.7.3a and quantified using featureCounts version 1.6.4. Values were normalized as reads per kilobase of exons per million mapped reads. We integrated RNA-seq data using ComBat-seq to remove batch effects [32]. Gene ontology (GO) analysis was performed using Database for Annotation, Visualization, and Integrated Discovery software 2021. Gene set enrichment analysis (GSEA) was performed using GSEApy version 0.9.9. The hallmark and C6 (oncogenic signature) gene sets were downloaded from the Molecular Signatures Database (version 2022.1.Hs: http://www.broadinstitute.org/gsea/index.jsp). Squamous PDAC and Progenitor PDAC identity signatures [33], and Basal-like and Classical signatures [15] have been previously described.
Analogy of genomic mutationsGenomic mutations such as SNPs and indels were inferred using the RNA-seq data. Somatic variant calling was performed as described previously [34]. Duplicate reads were flagged using Mark-Duplicates (Picard, version 2.27.4) and split into exons using SplitNCigarReads (GATK, version 4.2.6.1). Variant calling was performed using the Mutect2 (GATK, version 4.2.6.1).
Immunohistochemistry (IHC) and immunofluorescence (IF) stainingParaffin-embedded blocks of PDAC tissues were cut into 4 µm-thick sections and subjected to standard hematoxylin and eosin (H&E) staining and immunostaining. PDOs were isolated from Matrigel using Cell Recovery Solution (354,253; Corning) according to the manufacturer’s instructions. For IHC staining, sections were deparaffinized and rehydrated. Heat-mediated antigen retrieval was performed using sodium citrate buffer (pH6) for GATA6 and Tris/EDTA buffer (pH 9) for CK5. The sections were treated with 3% H2O2 in methanol to block endogenous peroxidase activity and then blocked with Blocking One Histo (Nacali Tesque, Kyoto, Japan). They were then washed with PBS/0.1% Triton X-100 and incubated with primary antibodies against GATA6 (AF1700; R&D SYSTEMS, Minneapolis, MN, USA; 10 μg/mL) and CK5 (GTX113219; GeneTex, Irvine, CA, USA; 1:1000) for 1 h at room temperature. Subsequently, the sections were incubated with the appropriate secondary antibody: horseradish peroxidase (HRP)-conjugated anti-goat IgG antibody (ab6741; Abcam, Cambridge, UK; 1:1000), anti-rabbit IgG (ab6721; Abcam; 1:1000), Alexa Fluor 488 anti-rabbit (ab150077; Abcam; 1:500), and 555 anti-goat (ab150130; Abcam; 1:500). Thereafter, 3,3'-diaminobenzidine substrate chromogen solution (11,209-1A; Kanto Kagaku, Tokyo, Japan) was applied, followed by counterstaining with hematoxylin. For IF staining, the nuclei were counterstained with 4',6-diamidino-2-phenylindole (Dojindo, Kumamoto, Japan).
Drug response assayThe PDOs were dissociated into single cells, and a 1000 live cells were seeded in a 96-well plate (B&W IsoPlate-96 TC; PerkinElmer, Kanagawa, Japan) containing Matrigel and complete medium. Therapeutic drugs, namely GEM (073-06631; FUJIFILM WAKO, Osaka, Japan; ranging from 1.0 × 10−10 to 1.0 × 10−5 mol/L), ulixertinib (BVD-523, HY-15816; MedChemExpress, NJ, USA; 1.0 × 10−6 mol/L), and chloroquine (HY-17589A; MedChemExpress, 5.0 × 10−6 mol/L) were added 24 h after plating and tested in triplicate. After 5 d, cell viability was quantified using the CellTiter-Glo 2.0 Kit following the manufacturer’s instructions (Promega, Madison WI, USA).
Statistical analysisAll statistical analyses were performed using Microsoft Excel, JMP Pro statistical software (ver. 16; SAS Institute Inc., Cary, NC, USA), and GraphPad Prism 9 software (Graphpad Software, Inc., Boston, MA, USA). Comparisons between two groups were assessed using Welch’s t test for continuous variables (normal distribution), Wilcoxon rank-sum test for continuous variables (non-normal distribution), or Fisher’s exact test for categorical data. Survival curves were analyzed using the Kaplan–Meier method with the log-rank test. Statistical significance was set at P < 0.05.
留言 (0)