
記住我



The grapevine (Vitis vinifera) phyllosphere is colonized by bacteria and fungi that substantially modulate grapevine health, fruit development and ripening, as well as the quality of grapes and wine (Barata et al., 2012; Martiniuk et al., 2023). The microbiome of a grapevine plant has direct and indirect relationships with its host and specific microbial biodiversity linked to a particular vineyard location is reported to be a crucial aspect, in conjunction with edaphic, climatic and human factors, in the concept of wine terroir (Gilbert et al., 2014). The concept of terroir is essential in viticulture since it links the sensory qualities of wine to the environmental conditions in which the grapes are grown (Van Leeuwen and Seguin, 2006).
Several scientific studies have shown that non-random biogeographic distribution patterns of microbial communities of grape surface in vineyards can be modulated by a combination of numerous factors, including geographical location, farming system, soil, cultivar, vintage and climate at various levels (Bokulich et al., 2014; Martins et al., 2014; Knight et al., 2015; Singh et al., 2018). These factors also affect the biology of the vines and the composition of the grapes, which in turn affect the microbiota, and, consequently, the qualitative properties of grapes and wine (Belda et al., 2017). This cycle has given rise to the concept of microbial-wine-terroir, intended as the close correlation between microbes, wine and territories (Gilbert et al., 2014; Knight et al., 2015).
To know the biogeographical distribution of vineyard microbial communities in specific regions, it is important to understand whether the vines themselves select different microorganisms according to their physiological responses, different environmental conditions and viticultural practices (Gilbert et al., 2014; Zarraonaindia et al., 2015; Pacifico et al., 2019; Vitulo et al., 2019). Therefore, in recent years, there has been a surge in the search for regional microbial signatures and microbial biogeography of wine and we now have a greater understanding of microbial diversity among wine-producing regions to begin to understand how this biodiversity can contribute to wine quality and regional characterization (Bokulich et al., 2014, 2016). Native yeasts, residing in a niche site, represent an important component of the microbiota of a vineyard, as they can influence the chemical and sensory profile and the so-called typicality of wine in a unique, reproducible, and identifiable way (Belda et al., 2017). Several studies have shown that the microbiota involved during the early fermentation stages, which is partially determined by endophytic plant-borne yeasts and bacteria, can comply with a well-delineated biogeography reflecting the signatures of different winegrowing regions with a minor influence from the grape variety and vintage year (Barata et al., 2012; Bokulich et al., 2014; Pretorius, 2020; Kamilari et al., 2021).
The diversity of the grape microbiota has long been studied using culture-dependent techniques (Barata et al., 2012; La Hens et al., 2014; Takahashi et al., 2014; Testa et al., 2014; Camilo et al., 2022), which have the disadvantage of being limited by the cultivability of microorganisms (Cocolin et al., 2013). Indeed, although these techniques are widely used for rapid and sensitive profiling of microbial communities, they are unable to detect a significant fraction of the microbial species present on grapes as rare taxa, low-abundance taxa and living but non-viable microorganisms (Bodor et al., 2020). The limitations of the conventional methods have led to the development of high-throughput sequencing (HTS)-based approaches to study the structure of microbial communities (Bleidorn, 2015). These innovative techniques allowed the scientific community to retrieve novel information about microbial communities in all different kinds of environments. As a result, the application of HTS methods for in-depth assessment of the grape and wine microbiome has increased in recent years (Salvetti et al., 2016; Morgan et al., 2017; Dissanayake et al., 2018; Kamilari et al., 2021). In a previous study we already assessed the impact of pedoclimatic conditions on the enological performance of two red cultivars grown throughout Southern Italy (Iorizzo et al., 2023). In this study, we obtained new information by adding other wine-grape samples of two cultivars (Cabernet and Aglianico) to determine relationships between spatial proximity and fungal composition through amplicons sequencing of the internal transcribed spacer (ITS2) region using a HTS approach combined with bioinformatics.
2 Materials and methods 2.1 Berry sample collection and processingGrape berries from cultivars Aglianico and Cabernet were collected in two different vineyards of Southern Italy (Sicily and Molise, 37° 40′; 400–700 m a.s.l. and 41° 42′; 606 m a.s.l., respectively) at the right maturation time, following the BBCH scheme described by Lorenz et al. (1995) (BBCH stage 89, berries ripe for harvest), during the 2020 growing season. Grape barriers from a third farm located in the Campania region (41° 13′; 100 m a.s.l.) and devoted to the production of Aglianico and Cabernet were also withdrawn in the same period. For each farm and for each cultivar, 30 clusters were harvested from different positions of the vineyard and from random positions on the plant to ensure the representation of the entire vineyard as previously described (Iorizzo et al., 2023). Briefly, at least ten berries were randomly selected from different parts of the cluster, avoiding those with visible damage and/or signs of pathogen infection, and pooled with berries from the other plants. For subsequent analyses, two different samples of 50 whole berries were taken and immediately transported to the laboratory, frozen in liquid nitrogen, and stored at −80°C for subsequent analyses.
2.2 Metataxonomic and bioinformatic analyses of grape fungal communitiesFungal community composition was analyzed by a culture-independent approach using next generation sequencing (NGS), as described in a previous study (Iorizzo et al., 2023). Total genomic DNA was extracted using the Stool DNA Isolation Kit (Norgen, Biotek Corp., Thorold, ON, Canada) according to the manufacturer’s instructions. Two replicates were produced for each sample. DNA was extracted from 200 mg of the homogenized sample. Extracted DNA was verified by agarose gel electrophoresis. The purity and amount of DNA was measured using a NanoDrop (NanoDrop 2000/2000c, Thermo Fisher Scientific, Italy) and standardized to a concentration of 10 ng/μl. The ITS2 region of the rRNA was amplified using primers 2024F and 2409R (White et al., 1990; Zhang et al., 2018). Amplicons were sequenced using the Illumina MiSeq PE300 platform (Illumina, San Diego, CA, USA) at Biodiversa s.r.l. (Rovereto, Italy). Raw paired-end reads obtained from NGS were demultiplexed using the QIIME 2 (ver. 2022.2) pipeline (Caporaso et al., 2010) and then were analyzed to evaluate the quality of sequencing with the DADA2 plugin (qiime dada2 denoise-paired) in QIIME2 (Callahan et al., 2016). For the quality control of reads, the Quality Score value of 25 was used as a threshold and the low-quality regions of sequences were removed. Reads were truncated at position 300 and 254 for the forward and reverse read, respectively. Moreover, to remove the base that belongs to the primer sequences, the first 5 nucleotides of reads were removed. At the end of DADA2 analysis, an amplicon sequence variant (ASV) table was obtained which recorded the frequencies of any ASV observed in each sample. The ASV table was filtered with the plugin qiime feature-table filter-features to discard features with frequency lower than 10 (0.001%).
ASVs were classified using the QIIME2 naïve Bayes classifier (qiime feature-classifier fit-classifier-naive-bayes), trained on the UNITE database (ver. 9.0, release 16-10-2022) (Abarenkov et al., 2022).
The new raw data of Campania samples were deposited in Mendeley Data with the accession number doi:10.17632/6jcyrwj2gh.
2.3 Statistical analysisStatistical analysis and graph processing were conducted on R (Version 4.2.3) using RStudio software (Version 2022.07.0). Several packages were used in the analysis: phyloseq (Version 1.42.0) to facilitate the import, storage, handling and analysis of the microbiome data (McMurdie and Holmes, 2013), as well as to determine alpha diversity. For the latter, both Chao1 and Shannon indexes were calculated. Vegan (Version 2.6-4) package was used for the permutational multivariate analysis of variance (PERMANOVA) using the Bray Curtis metric distance. The mixOmics package was used for sparse partial least squares discriminant analysis (Rohart et al., 2017) and the microbiomeMarker (Version 1.4) package for the Linear Discriminant Analysis (LDA) effect size (SE). The ggplot2 (Version 3.4.4) package was used for graphical representation of the data.
3 Results 3.1 Bioinformatic analysisIn the year 2020 two biological replicate samples of grapes were collected from two vineyards (Aglianico and Cabernet) located in Southern Italy (Sicily, Campania and Molise regions, Supplementary Table 1). Surface fungi community composition of grapes was analyzed through high-throughput amplicon sequencing of the ITS2 region.
A total of 923,559 paired-end sequences were obtained from sample sequencing. The average of reads per sample was 76,963, ranging from 54,059 to 123,628. After reads quality check, denoising and chimera filtering (Supplementary Figure 1), 465,805 paired-end sequences were obtained (Supplementary Table 2) corresponding to 419 ASVs. The ASVs were filtered with the aim to discard features with frequency lower than 10. At the end of bioinformatic analysis, a feature table of 465,469 reads, corresponding to 323 ASVs, was obtained and used for the subsequent analysis. The rarefaction curves reached the plateau for all assayed samples, suggesting that the sequencing depth was appropriate (Supplementary Figure 2).
3.2 Grape fungi community profileFrom the taxonomy classification 3 phyla, 17 classes, 45 orders, 96 families, 122 genera and 172 species were identified. In general, the analysis of fungal community confirmed our previous findings (Iorizzo et al., 2023). In fact, Ascomycota remained the most abundant phylum, representing 87.9% ± 9.4% of total reads, followed by Basidiomycota 11.8% ± 9.3% and Chytridiomycota 0.02% ± 0.08% in all analyzed samples. The remaining 0.3% ± 0.2% of the reads were represented by unclassified fungi (Figure 1A and Supplementary Table 3).
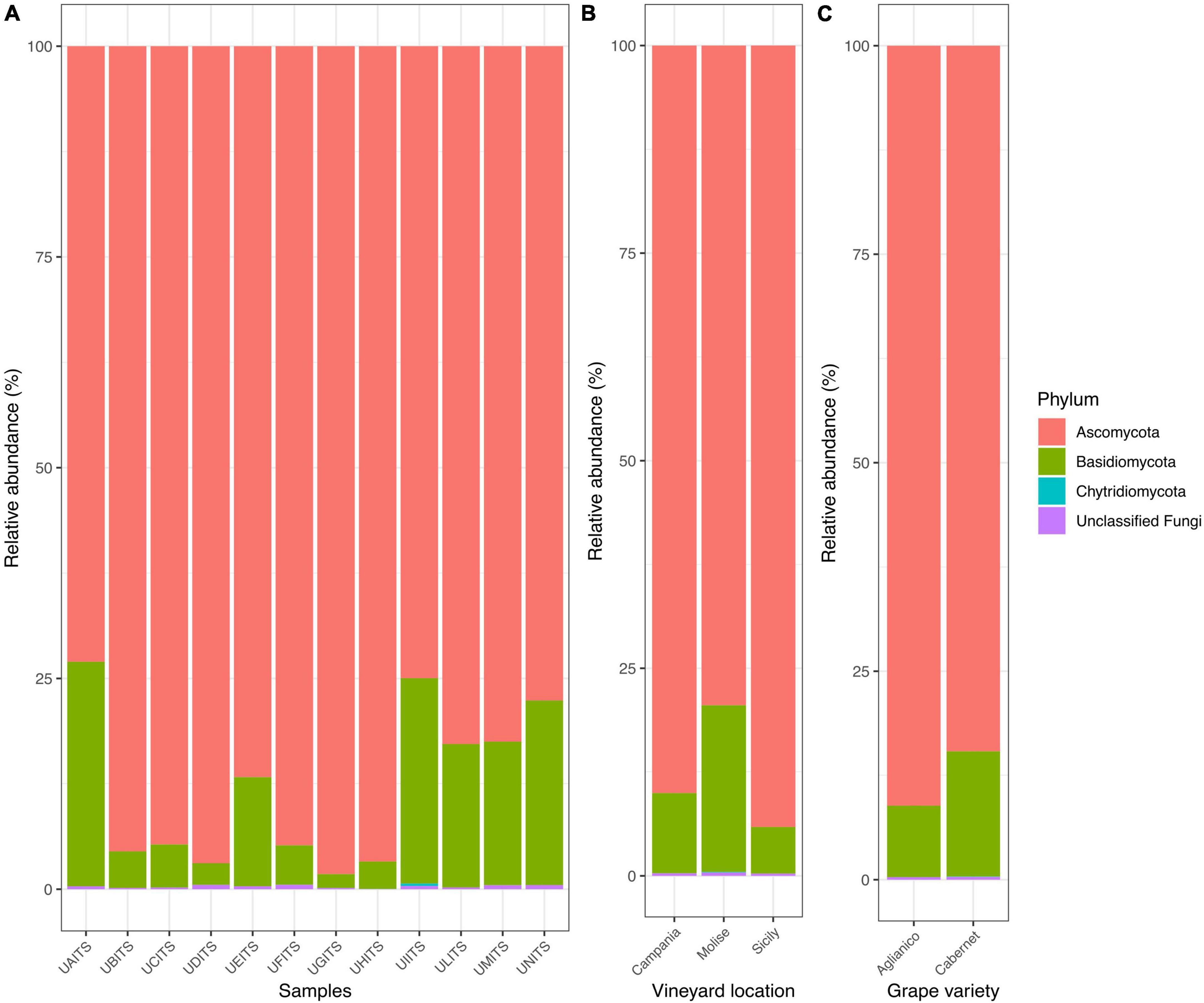
Figure 1. Stacked bar plot illustrates the distribution of the relative abundance of the grape fungi community at the phylum level. The relative abundance is reported for panel (A) single samples, (B) samples grouped by vineyard location (VL), and (C) samples grouped by grape variety (GV). The sample codes are specified as follow: UAITS, Cabernet_Campania_1; UBITS, Cabernet_Campania_2; UCITS, Aglianico_Campania_2; UDITS, Aglianico_Campania_3; UEITS, Cabernet_Sicily_1; UFITS, Cabernet_Sicily_3; UGITS, Aglianico_Sicily_1; UHITS, Aglianico_Sicily_2; UIITS, Cabernet_Molise_2; ULITS, Cabernet_Molise_3; UMITS, Aglianico_Molise_1; UNITS, Aglianico_Molise_2. See Supplementary Table 1 for more details.
The new data processed in this study considered two variables: vineyard location (VL) and grape variety (GV). Regarding the results of relative abundance with respect to the variable VL (Figure 1B and Supplementary Table 4), we found that the grape samples from Campanian vineyard were characterized by 90% of reads belonging to the Ascomycota and about 9.7% of reads were referable to the Basidiomycota. Similarly, in the grape samples from Sicilian vineyard, an average of 94% of the reads were associated with the phylum Ascomycota and only 5.6% of them belonged to the Basidiomycota. A slightly different result was found in the Molisian samples as Ascomycota accounted for around 74% of the reads, while around 20% of the reads belonged to Basidiomycota. When the samples were grouped according to the GV (Figure 1C and Supplementary Table 5), we found that Ascomycota represented 91.1 and 84.6% of the phyla for the Aglianico and Cabernet cultivars, respectively. Basidiomycota, on the other hand, were around 8.6% in the Aglianico samples and 15% in the Cabernet samples.
Regarding the classification at the family level, out of 96 families identified, only 9 of them enclosed about 95% of total reads. In detail, Cladosporiaceae was the most abundant family, representing an average of 28.4% ± 13.5 of the total reads, followed by Saccotheciaceae 24.8% ± 14.0%, Pleosporaceae 15.9% ± 8.3%, Saccharomycodaceae 12.3% ± 18.2%, Sporidiobolaceae 7.40% ± 8.80%, Didymellaceae 2.7% ± 4.5%, Filobasidiaceae 1.70% ± 1.53%, Bulleribasidiaceae 1.2% ± 1.9%, and Saccharomycetaceae 0.9% ± 0.9% (Figure 2A and Supplementary Table 6). With regards to the distribution of the 9 most abundant families according to the VL (Figure 2B and Supplementary Table 7), we observed that Cladosporiaceae were present with a prevalence of about 20.6, 34.7, and 29.9% in vineyards of Campania, Molise and Sicily regions, respectively. Saccotheciaceae showed a prevalence of 33.6% in samples from Campanian vineyard, 11.7% in Molisian vineyard and 29% in Sicilian vineyard. Reads associated with Pleosporaceae were present at levels of 17.4, 22.1, and 8.3% in samples from vineyards located in Campania, Molise and Sicily regions, respectively. The results regarding the distribution of Saccharomycodaceae among the samples were particularly interesting. In fact, a consistent presence of reads attributable to Saccharomycodaceae were detected only in Campanian (14.4%) and Sicilian (22.1%) vineyards. On the contrary, only 0.4% of Saccharomycodaceae were observed in Molisian vineyard. Sporidiobolaceae were detected at relevant presence in Campanian (6.9% ± 10.9%) and Molisian (15.2% ± 3.4%) vineyards, while their presence on grapes from Sicilian farm was only 0.1%. Another interesting result concerns the Didymellaceae family. In this regard, a consistent presence of reads was observed only in Molisian vineyard (7.2%), while a poor relative abundance was detected in the other samples (0.6% Campania and 0.4% Sicily).
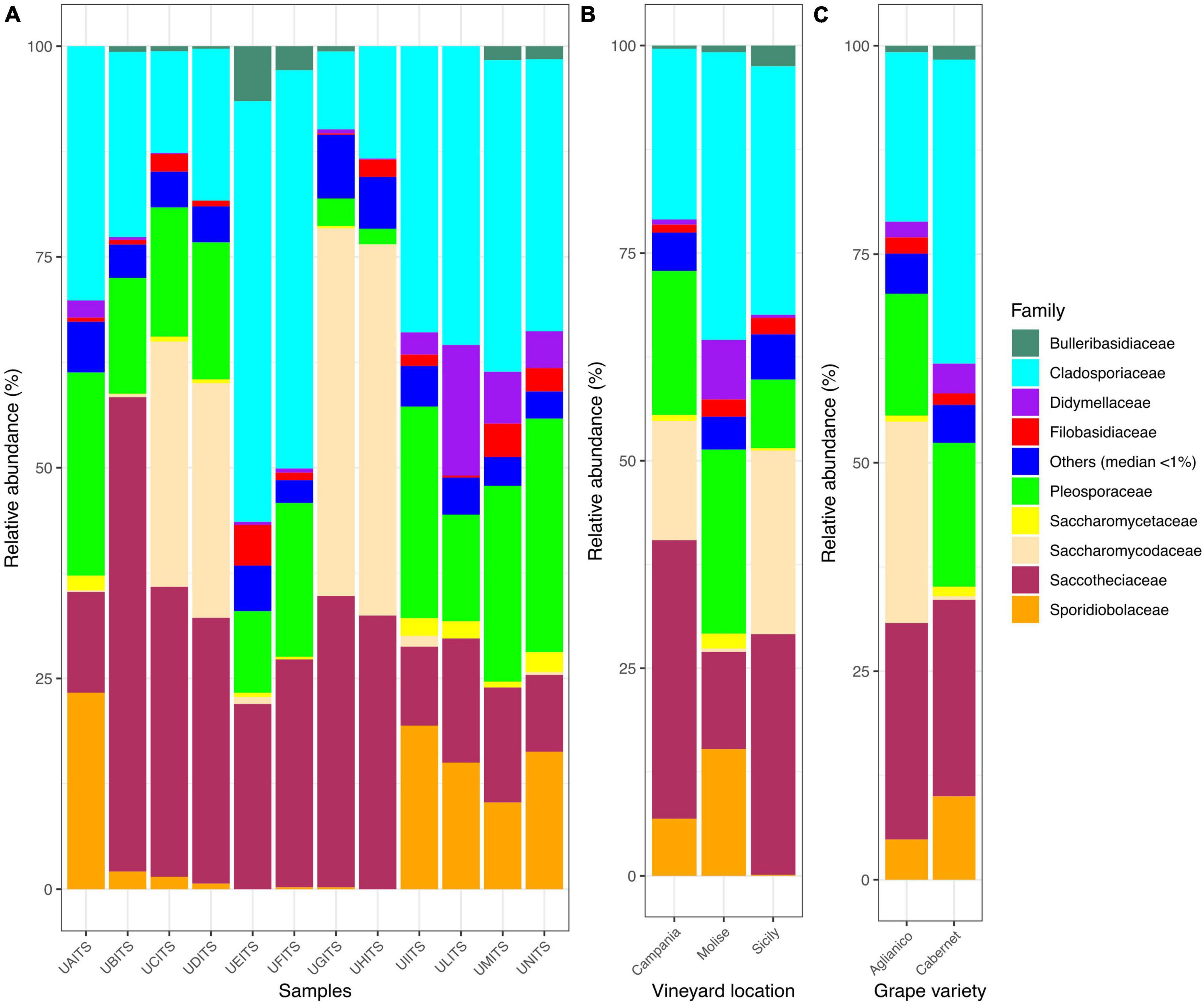
Figure 2. Stacked bar plot illustrates the distribution of the relative abundance of the grape fungi community at the family level. The group indicated as “Others (median < 1%)” encompasses families that show a relative abundance below median value 1. The relative abundance is reported for panel (A) single samples, (B) samples grouped by vineyard location (VL), and (C) samples grouped by grape variety (GV). The sample codes are specified as follow: UAITS, Cabernet_Campania_1; UBITS, Cabernet_Campania_2; UCITS, Aglianico_Campania_2; UDITS, Aglianico_Campania_3; UEITS, Cabernet_Sicily_1; UFITS, Cabernet_Sicily_3; UGITS, Aglianico_Sicily_1; UHITS, Aglianico_Sicily_2; UIITS, Cabernet_Molise_2; ULITS, Cabernet_Molise_3; UMITS, Aglianico_Molise_1; UNITS, Aglianico_Molise_2. See Supplementary Table 1 for more details.
Investigating the distribution of the nine families in relation to the variable VL (Figure 2C and Supplementary Table 8), we observed that Saccharomycodaceae were particularly abundant (24.1%) in Aglianico grapevine and almost undetectable in Cabernet samples (0.4%).
In relation to genera classification, out of 122 genera detected, only 10 groups were present in almost all samples (Figure 3A and Supplementary Table 9). In detail, Cladosporium was present with an average of 28.3%, followed by Aureobasidium (24.8%), Alternaria (13.5%), Hanseniaspora (12.3%), Sporobolomyces (7.3%), unclassified Didymellaceae (2.7%), Stemphylium (2.3%) Filobasidium (1.7%), Vishniacozyma (1.1%) and Saccharomyces (0.9%). Genera grouped as “Others (median < 1)” showed about 5% of average relative abundance. Grouping the genera distribution according to the VL variable (Figure 3B and Supplementary Table 10), it was observed that in Campanian vineyard the genus Aureobasidium was the most abundant (33.5%), followed by Cladosporium (20.5%), Hanseniaspora (14.4%), and Alternaria (14.3%). Samples from Molise vineyard were also characterized by the high presence of Cladosporium (34.4%), Alternaria (18.5%) and Aureobasidium (11.7) genera, but contrary to the Campania samples, a high amount of Sporobolomyces (15.2%) and a low presence of Hanseniaspora (0.4%) was observed. With regard to the samples from Sicilian vineyard, Cladosporium (29.8%), Aureobasidium (29.0%) and Hanseniaspora (22.1%) were the most abundant genera, instead Alternaria (7.5%) and Sporobolomyces (0.1%) were present in a small number. When the dataset was stratified according to the GV variable (Figure 3C and Supplementary Table 11), Cladosporium was the most abundant genus (34.3%) in the Cabernet grape variety, followed by Aureobasidium (18.3%) and Alternaria (14.6%), while in the Aglianico samples, a significant amount of reads attributable to the genus Hanseniaspora (28.4%) was recorded in addition to a high presence of Aureobasidium (32.0%), Cladosporium (15.7%) and Alternaria (14.6%).
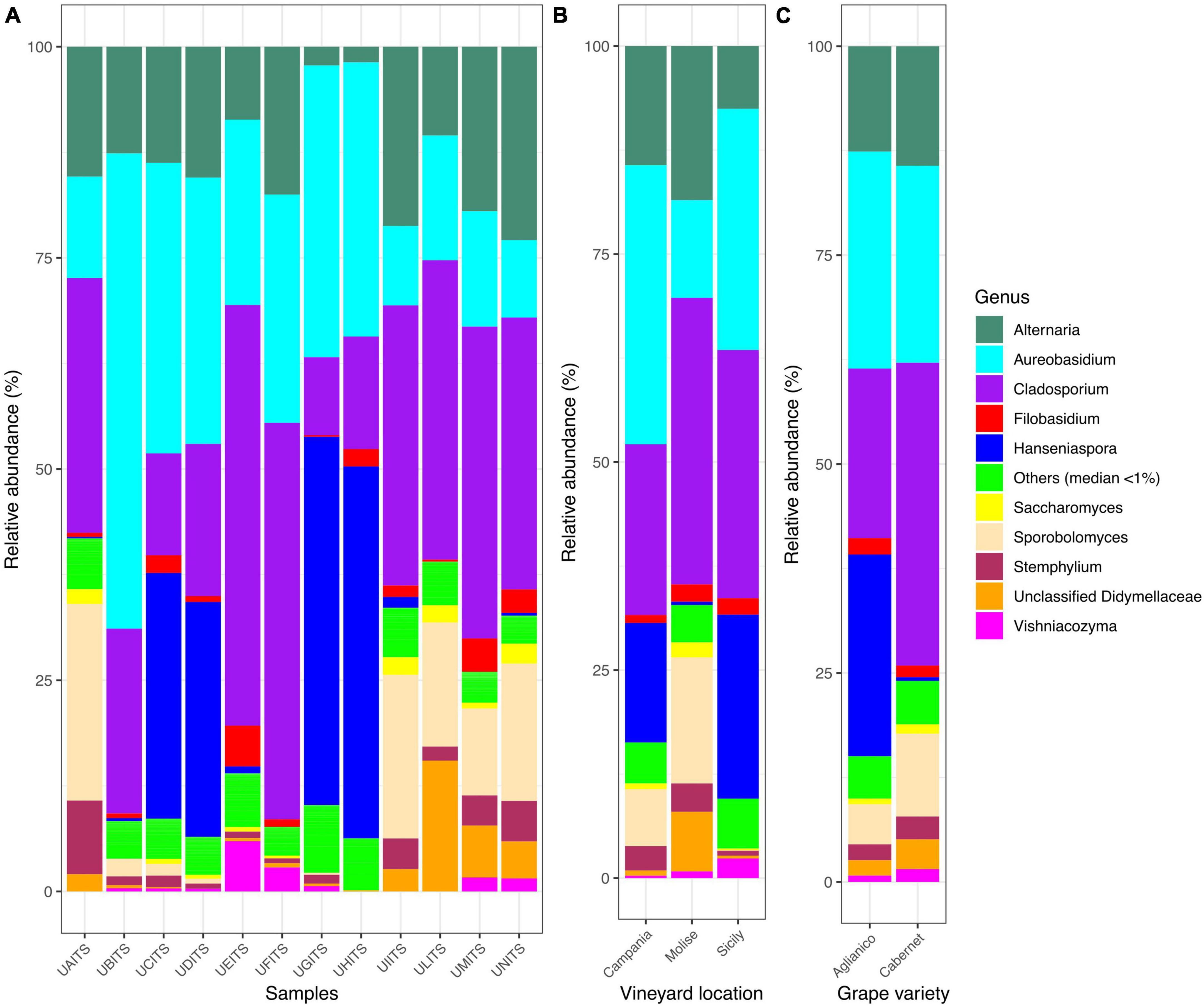
Figure 3. Stacked bar plot illustrates the distribution of the relative abundance of the grape fungi community at the genus level. The group indicated as “Others (median < 1%)” encompasses genera that show a relative abundance below median value 1. The relative abundance is reported for panel (A) single samples, (B) samples grouped by vineyard location (VL), and (C) samples grouped by grape variety (GV). The sample codes are specified as follow: UAITS, Cabernet_Campania_1; UBITS, Cabernet_Campania_2; UCITS, Aglianico_Campania_2; UDITS, Aglianico_Campania_3; UEITS, Cabernet_Sicily_1; UFITS, Cabernet_Sicily_3; UGITS, Aglianico_Sicily_1; UHITS, Aglianico_Sicily_2; UIITS, Cabernet_Molise_2; ULITS, Cabernet_Molise_3; UMITS, Aglianico_Molise_1; UNITS, Aglianico_Molise_2. See Supplementary Table 1 for more details.
3.3 Alpha diversity of grapevine according to the vineyard location and grape varietyThe estimation of intragroup diversity (alpha diversity) was carried out to verify the hypothesis that the fungi richness and biodiversity vary within the VL (Campania, Molise, and Sicily) and the GV (Aglianico and Cabernet). For this purpose, Chao1 index was used to estimate the species richness and the Shannon index was used to estimate the diversity. Results are reported in Figure 4A and show that there were no significant differences (Anova test, p = 0.516) in richness (Chao1) between GV in the three vineyards. In contrast, a significant difference (Anova test, p = 0.021) of diversity (Shannon) between VL was found. Specifically, the post hoc test showed that the Molisian vineyard was characterized by a high diversity (TukeyHSD test, p = 0.018) compared to the Sicilian one. No significant differences (TukeyHSD test, p > 0.05) were found between vineyards from Molise and Campania regions or between Campania and Sicily regions. Regarding the alpha diversity calculated from the two GV, results showed that there were no significant differences (t-test, p > 0.05) for species richness and diversity (Figure 4B).
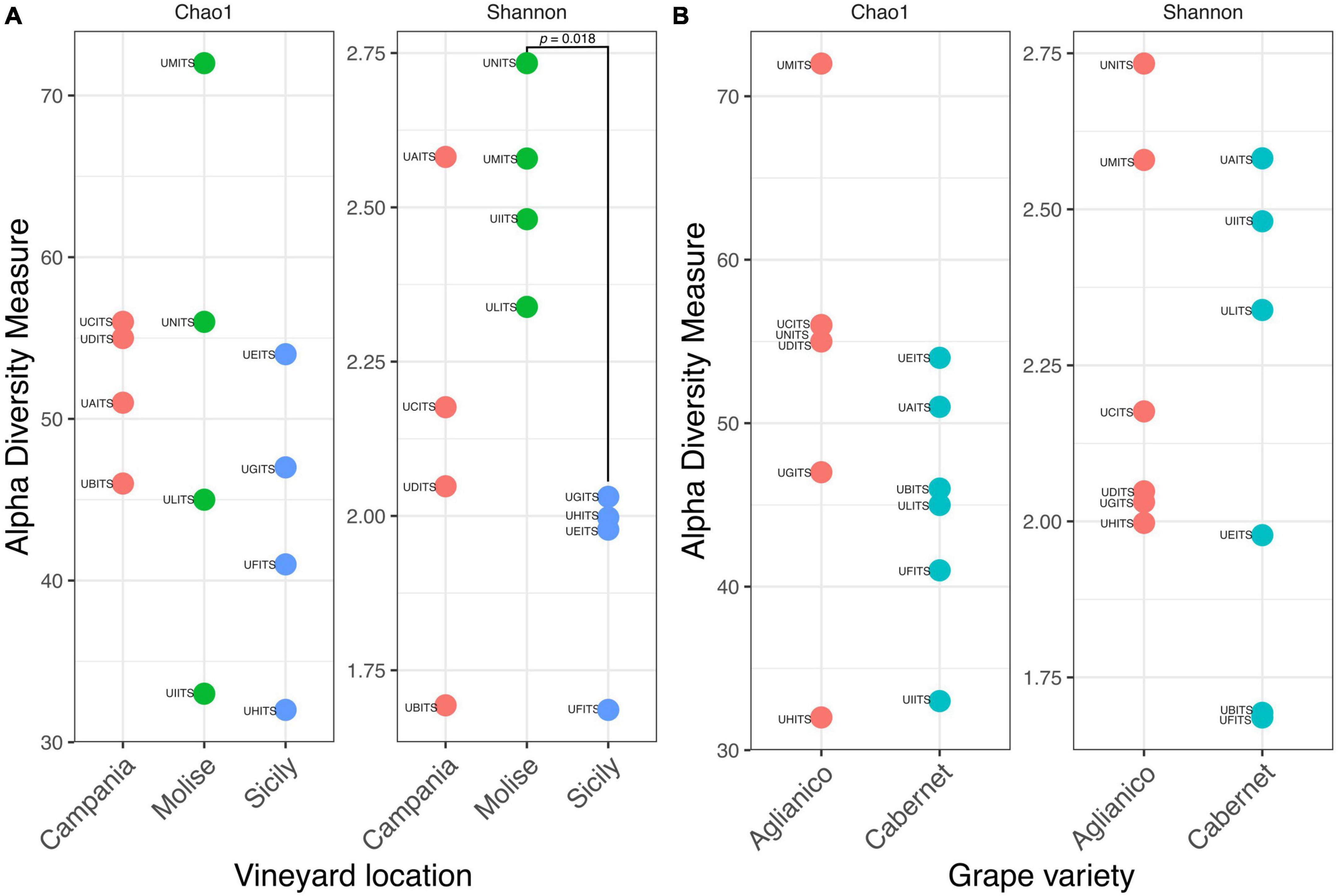
Figure 4. Alpha diversity of grapes calculated according to the variables (A) vineyard location (VL) and (B) grape variety (GV). Species richness and diversity were calculated with Chao1 and Shannon indexes, respectively. The sample codes are specified as follow: UAITS, Cabernet_Campania_1; UBITS, Cabernet_Campania_2; UCITS, Aglianico_Campania_2; UDITS, Aglianico_Campania_3; UEITS, Cabernet_Sicily_1; UFITS, Cabernet_Sicily_3; UGITS, Aglianico_Sicily_1; UHITS, Aglianico_Sicily_2; UIITS, Cabernet_Molise_2; ULITS, Cabernet_Molise_3; UMITS, Aglianico_Molise_1; UNITS, Aglianico_Molise_2. See Supplementary Table 1 for more details.
3.4 Beta diversityBeta diversity based on Bray Curtis distance was calculated to evaluate the degree of variation in species composition among the samples. Results showed that the fungal population segregated mainly according to the VL and slightly according to the GV (Figure 5). In detail, the PCoA plot shows a good separation of samples along axis 1 (explaining 47% of variation), while a less pronounced separation was observed along axis 2 (explaining 22% of variation). The effect and the significance of the variable VL (Campanian, Molisian and Sicilian vineyards) and GV (Aglianico and Cabernet) on the fungi was investigated through the Permutational Multivariate Analysis of Variance (PERMANOVA). The results revealed that the variable VL significantly (p = 0.0001) affected fungal communities, while the variable GV had no significant effect (p > 0.05). However, the interaction VL: GV showed a significant (p = 0.001) effect on the overall variance of the dataset (Supplementary Table 12). From multiple comparison tests, it was highlighted that samples from Molise vineyard were significantly different (p = 0.039) from those of Campanian and Sicilian vineyards. No differences (p > 0.05) were found between Campania and Sicily vineyards.
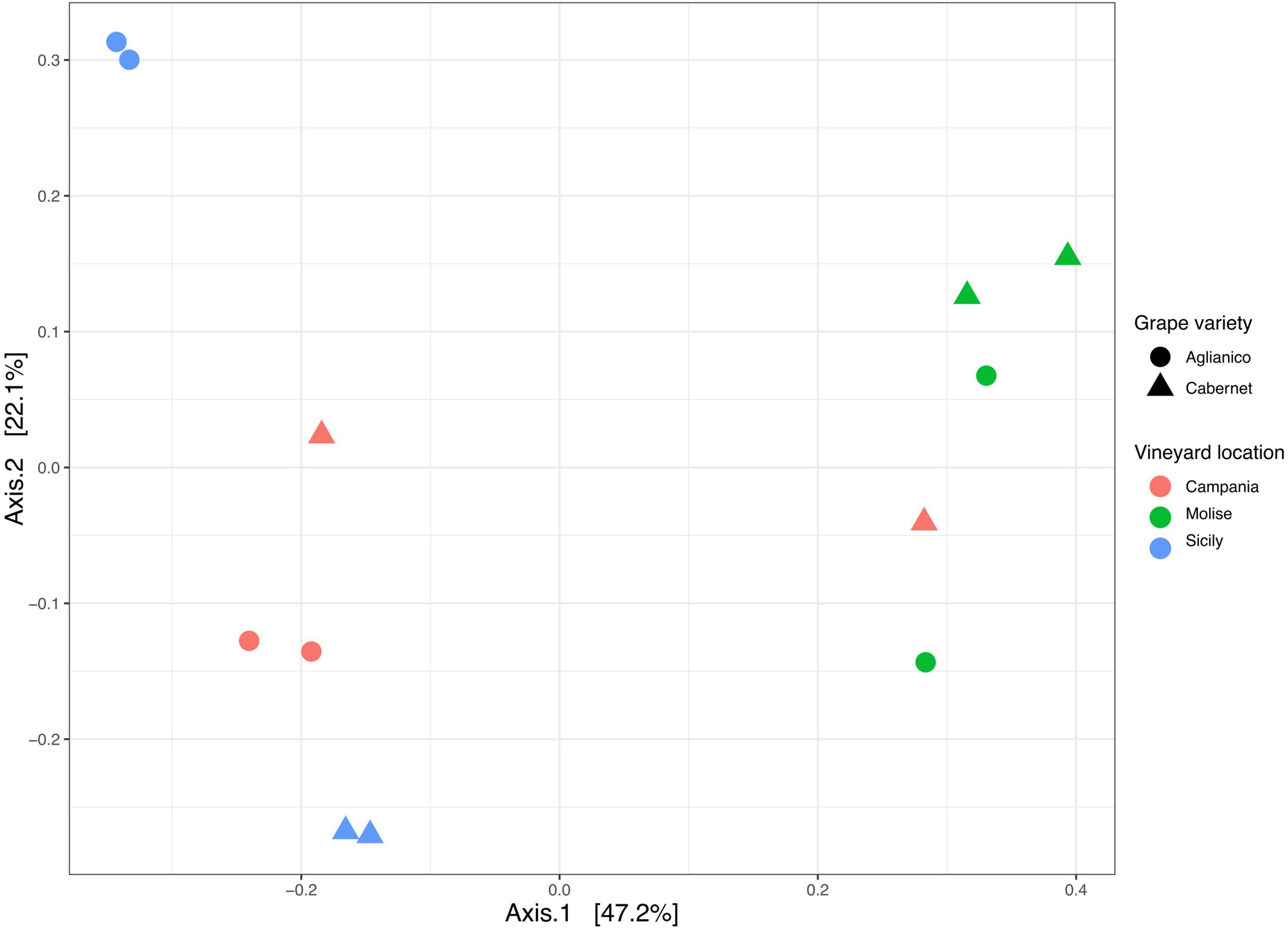
Figure 5. Principal Coordinate Analysis (PCoA) based on Bray Curtis distance generated with ASVs (at 99% identity) present in samples grouped according to vineyard location (VL) and grape variety (GV).
3.5 Discriminant analysisThe taxonomic groups driving differences between fungal communities were investigated with the sparse Partial Least Squares Discriminant Analysis (sPLS-DA). The results show that the variable VL influences the entire community (Figure 6). Grape samples from Molise vineyard separated from those of Sicilian vineyard along component 1 (X-variate 1), while grape samples from Campania vineyard separated from those of the other vineyards along component 2 (X-variate 2).
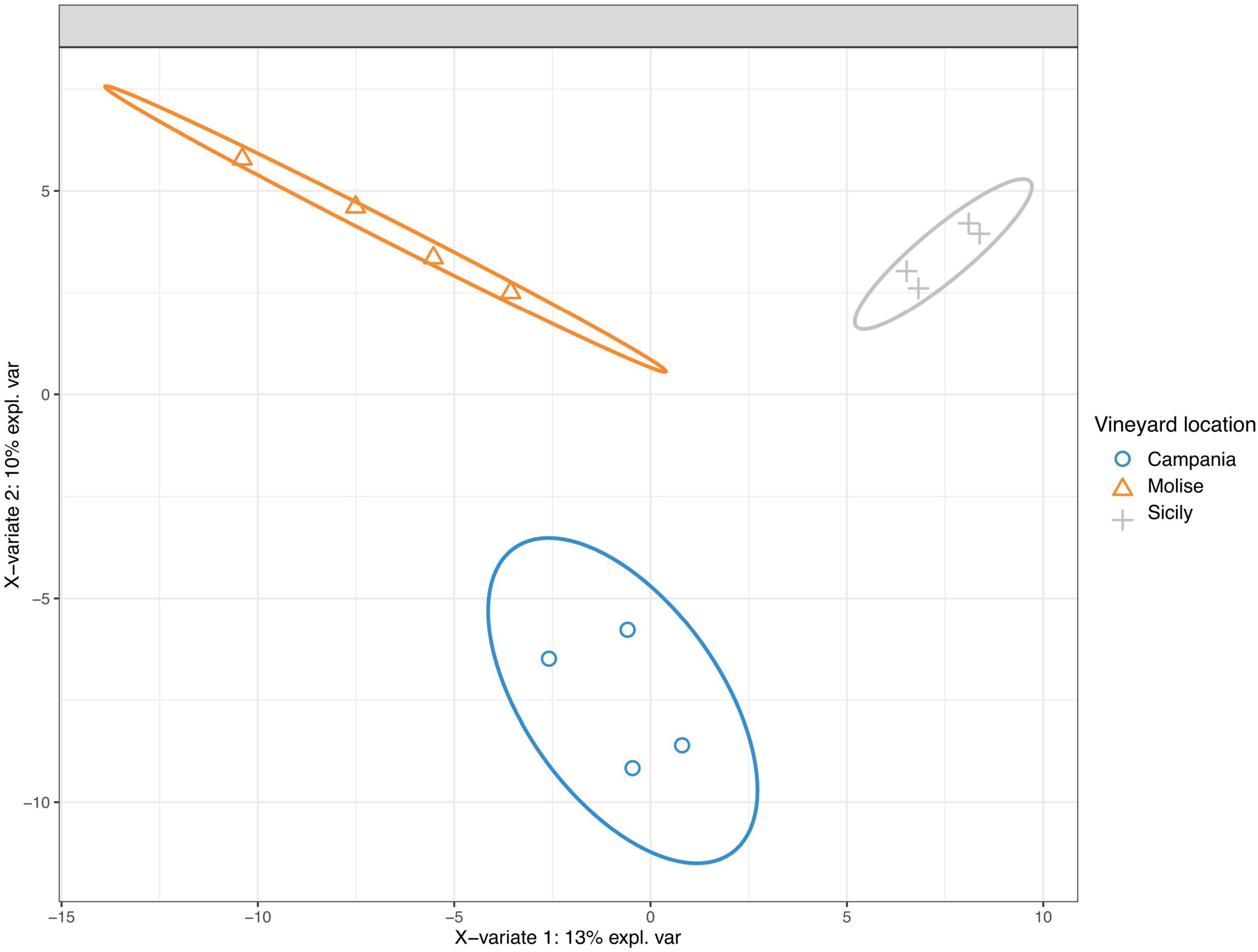
Figure 6. Sparse partial least-squares discriminant analysis (sPLS-DA) of read counts transformed using cumulative sum scaling normalization at the sequence variant level. The ellipse shows the 95% confidence level.
Taxa contributions on the first 2 components are reported in Figures 7A, B. Fungi that mainly increased in abundance in samples from Molise vineyard included Sporobolomyces roseus, Alternaria metacromatica, and Cladosporium herbarum, whilst fungi associated to samples from Sicilian vineyard belonged to the genus Hanseniaspora. Regarding taxa associated to Campanian vineyard, sPLS-DA analysis reported Phaeosphaeriaceae family and Pichia terricola, Zygoascus meyerae, Papiliotrema flavescens and Hanseniaspora spp. as the top 5 most abundant groups.
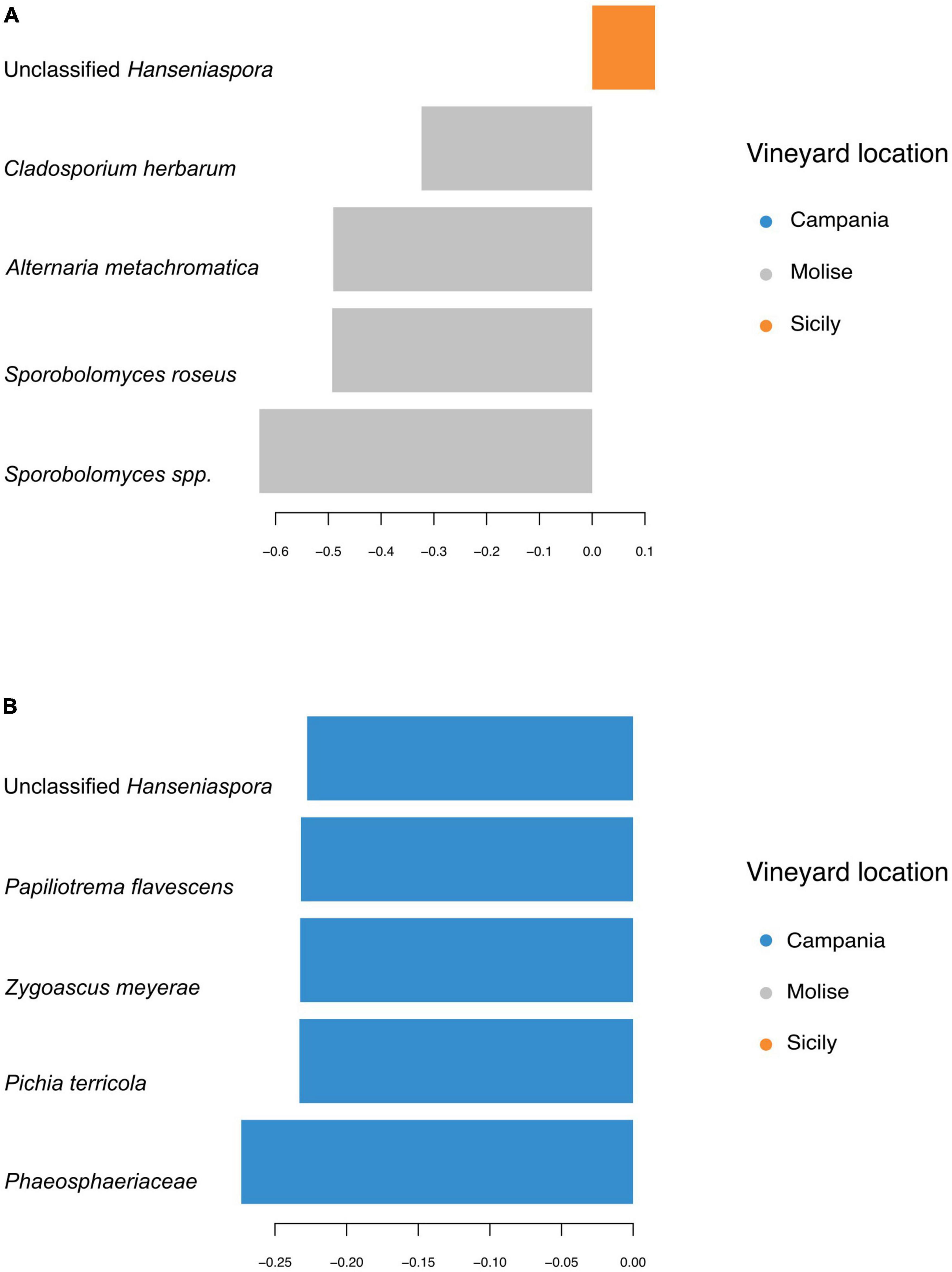
Figure 7. First five sequence variants contributing to the separation along with panel (A) component 1, and (B) component 2 of sPLS-DA from Figure 6. Bar length indicates loading coefficient ranked by importance, bottom to top; bar color indicates the group in which the sequence variant has the highest median abundance.
Overall, the results obtained from sPLS-DA were supported by the Linear discriminant analysis (LDA) effect size (LEfSe). LEfSe results revealed that 20 fungal taxa were mainly present in the samples of the three vineyards under study with significant differences (p < 0.05) at a threshold value of 1.5. In detail, one fungal taxon was associated with the Sicilian vineyard, three fungal taxa with the Campanian vineyard and 16 fungal taxa were significantly associated with the Molisian vineyard (Figure 8). In particular, according to results obtained by sPLS-DA analysis, the Phaeosphaeriaceae family was associated with the vineyard located in Campania region, while Phaeosphaeria caricicola, that resulted also associated with Campanian samples though LEfSe analysis, was not confirmed with sPLS-DA analysis. Regarding the samples belonging to the Molise vineyard, both LEfSe and sPLS-DA analysis confirmed Cladosporium herbarum, Sporobolomyces roseus, and Alternaria metachromatica significantly correlated to VL. Instead, Didymellaceae family, Sthemphylium genus, Saccharomycetaceae family and Pyrenophora genus resulted correlated to the Molise vineyard only when LEfSe analysis was applied. With regard to samples belonging to the Sicilian vineyard, LEfSe analysis reported that only Leotiomycetes was associated with the VL, contrarily to what it was obtained from the sPLS-DA analysis.
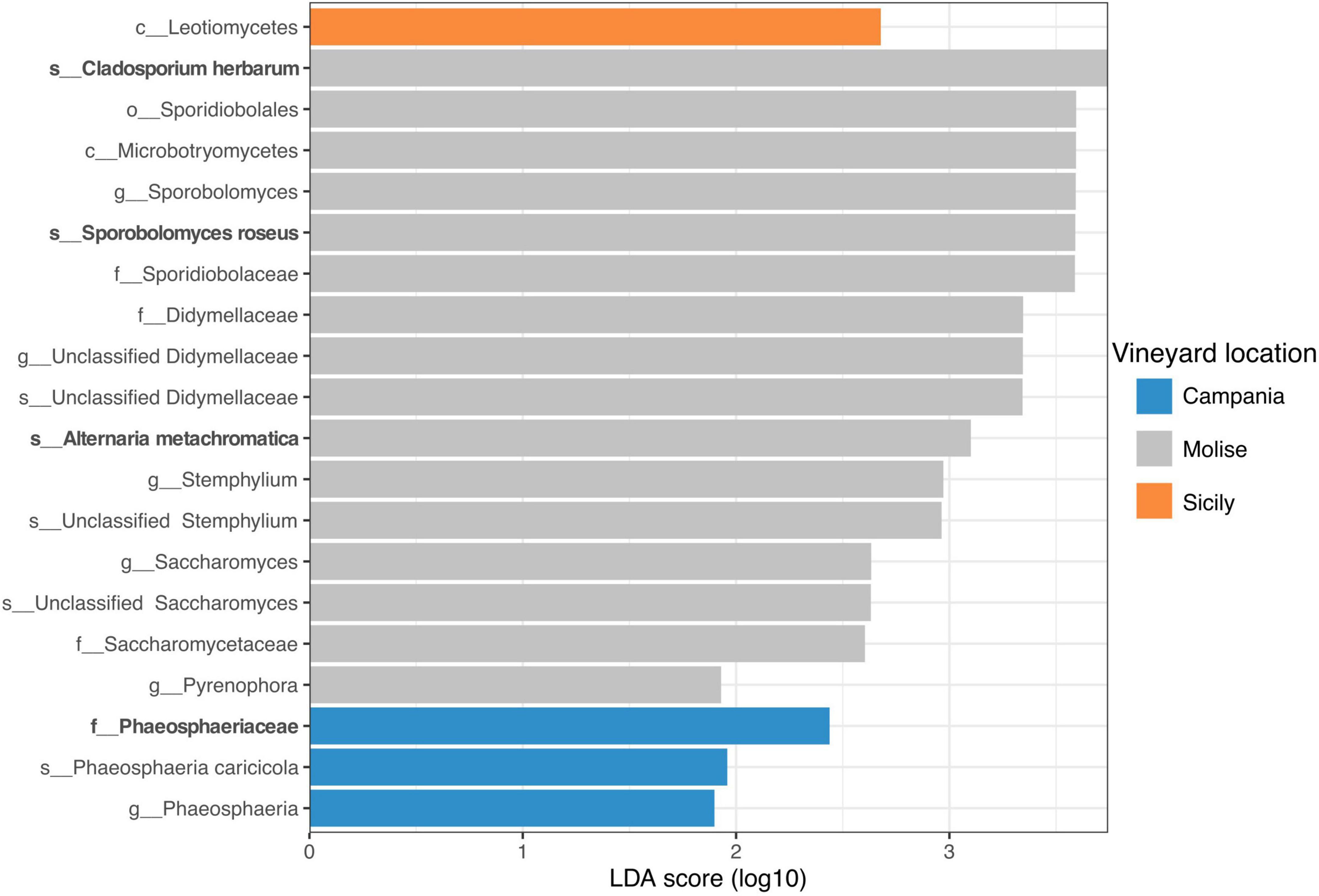
Figure 8. Linear discriminant analysis (LDA) effect size (LEfSe). Horizontal bars represent the effect size for each taxon. The cut off on the logarithmic LDA score for discriminative features was set to 1.5. The change of taxon relative abundance was statistically significant at p < 0.05. The name of the taxon level is abbreviated as p__phylum; c__class; o__order; f__family, and g__genus. Taxa name in bold were confirmed by sPLS-DA analysis reported in Figure 7.
4 DiscussionMicroorganisms have a key role in the winemaking process as they drive fermentation and are able to influence the aroma and quality of wine (Verginer et al., 2010; Belda et al., 2017; Sherman et al., 2020). Grape berries are one of the main sources of microbial communities that affect the winemaking process, therefore the study of the microbial diversity of wine grapes is of particular interest (Morrison-Whittle and Goddard, 2018). The structure of microbial communities could be, in turn, influenced by several factors such as geographical location, climatic conditions, grape variety, viticultural practices (e.g., fungicides, herbicides, fertilizers) and many others (Bokulich et al., 2014; Pinto et al., 2014; Setati et al., 2015; Grangeteau et al., 2017; Vitulo et al., 2019). As far as wine is concerned, individual vineyards are the smallest scale through which wine diversity can be studied (Knight et al., 2020) and understanding the scale at which microbial communities differ could be important in clarifying the concept of terroir.
Due to the complexity of the microbial population found on grape berries, it is evident that traditional culture-dependent methods have limitations in studying microbial diversity. On the contrary, high-throughput sequencing (HTS), coupled with multivariate data analysis, represents a valid method for studying microorganisms in a variety of contexts. In the present study, we used both HTS and multivariate data analysis to investigate the fungi community structure of grape samples collected in three different vineyards located in Campania, Molise and Sicily regions (Southern Italy) and belonging to two red grape varieties (Aglianico and Cabernet). In general, the descriptive approach through relative abundance analysis confirmed what we already found in our previous study (Iorizzo et al., 2023), but in the present research, new information is derived from the addition of new samples from the Campanian vineyard. Moreover, the statistical approach used furnished new important information on the structure of fungal communities as related to vineyard location and grape variety.
In detail, we found that the main phyla identified on grape berries were represented by Ascomycota, Basidiomycota and Chytridiomycota. In particular, the most abundant phylum in our samples was Ascomycota (∼ 88%), followed by Basidiomycota (∼ 12%). Similarly, Gao et al. (2019) reported that Ascomycota, Basidiomycota and Zygomycota were the main phyla found on the surface of grape berries from six wine regions in Xinjiang - China. In addition, other authors have also found Ascomycota and Basidiomycota as the dominant fungal phyla on grape berries (Morrison-Whittle et al., 2017). Recently, Xu et al. (2022) conducted a study in which they reported that Ascomycota and Basidiomycota were the dominant fungi present on wine-grape surfaces.
Regarding the fungal community structure at the family level, we showed that Cladosporiaceae, Saccotheciaceae, Pleosporaceae, Saccharomycodaceae and Didymellaceae were the most representative families within the phylum Ascomycota, while Sporidiobolaceae, Filobasidiaceae and Bulleribasidiaceae were the most abundant ones among the phylum Basidiomycota. Similarly, in previous studies, other authors reported that Pleosporaceae and several members of Saccotheciaceae and Cladosporiaceae were the dominant fungi in Riesling grape berries (Kecskeméti et al., 2016). Interestingly, stratifying the dataset according to the variable VL, we observed that Cladosporiaceae, Saccotheciaceae, Pleosporaceae, and Filobasidiaceae were present in all samples (n = 12), suggesting that these 4 families represent the core taxa. Similar results were also obtained by Gao et al. (2019) in their work regarding the study of the fungal community structure of grape surfaces. In addition, Wei et al. (2022) reported that the core taxa of Cabernet Sauvignon grapes are retained at the genus level, although the fungi diversity changes during the berry development.
Studying the fungal community structure of grape berries at genus level, on a dataset stratified in accordance with the variable VL, we found that the main genera (about 71% of OTUs) were represented by Cladosporium, Aureobasidium, Alternaria, Stemphylium, and Filobasidium. Similarly, when the fungal community structure of grape berries was studied on a dataset stratified by the variable GV, it was observed that, apart from the genus Stemphylium, all other genera were conserved, indicating that Cladosporium, Aureobasidium, Alternaria, and Filobasidium constitute the core taxon. Some of these genera were also reported as the most common fungal organisms isolated from grapes in other parts of the world. For example, in a study conducted in different regions of Portugal, the authors reported that Cladosporium, Alternaria, and Aureobasidium were the main genera of Alvarinho grapes (Fernandes et al., 2023). In another recent work conducted in the Xinjiang region of China, the authors found that the dominant fungi on nine grape skins variety were represented by Alternaria, Filobasidium, Aureobasidium, Erysiphe and Naganishia (Xu et al., 2022). In Italy, Perpetuini et al. (2022) reported that the common genera of fungi detected in grape samples of Montepulciano d’Abruzzo cultivar (Abruzzo Region, Central Italy) were represented by Hanseniaspora, Aureobasidium, Botrytis, Pichia, Cladosporium and Alternaria. In another work conducted by Perpetuini et al. (2023), it was found that Aureobasidium, Metschnikowia, Hanseniaspora, Botrytis, Cladosporium, Pichia, Alternaria, Epicoccum, Vishniacozyma and Candida were the main genera detected on the minor grapevine cultivar cv. Nero Antico from Chieti (Abruzzo Region, Central Italy). Similarly, Rossetti et al. (2023) studied the fungal community associated with conventional and organic Trebbiano Abruzzese grapes from vineyards of Chieti (Abruzzo Region, Central Italy), and a core taxon of 8 genera was detected (Zygosaccharomyces spp., Cladosporium spp., Botrytis spp., Hanseniaspora spp., Pichia spp., Alternaria spp., Candida spp., Aureobasidium spp.) in all samples investigated.
For a better understanding of the fungal community structure of grape samples and to assess the hypothesis that the fungal population of grape berries differs between samples depending on vineyard location(VL) and/or grape variety (GV), we used several statistical tools based on both univariate and multivariate approaches.
In detail, the alpha diversity values indicated that there were no significant differences (p > 0.05) in terms of richness (Chao1) between the analyzed samples, highlighting that the number of species present in the samples was essentially the same, regardless the VL and the GV (Figure 4). On the other hand, alpha diversity as measured with the Shannon index, evidenced that the VL may have a significant effect on the fungal community composition, as grapes from Molisian vineyard have a greater diversity than grapes from Sicilian one (Figure 4). This result indicates that there is a greater presence of rare species in grape samples from Molisian vineyard than those observed in Sicilian samples.
The effect of VL on the diversity of fungal communities among samples was also supported by the beta diversity measurement. In fact, both PCoA and sPLS-DA analyses showed that samples are mainly grouped according to the VL (Figure 5). These results are consistent with those reported in other studies where it was demonstrated that the diversity of the microbial community is influenced by geographical location of vineyards (Bokulich et al., 2014; Mezzasalma et al., 2017; Kioroglou et al., 2019; Papadopoulou et al., 2022; Kazou et al., 2023). Similar results were also obtained by other authors who have reported that VL has a greater impact on the fungal community than the state of ripeness (Kioroglou et al., 2019).
In the present study, a strong association between some families genera or species and their VL was confirmed by the results obtained using both sPLS-DA and LEfSe. For instance, both sPLS-DA and LEfSe analyses evidenced that C. herbarum, S. roseus and A. methacromatica were associated to Molisian vineyard. Similarly, the Campanian vineyard showed a strong association with the Phaeosphaeriaceae family, while they were weakly associated with Pichia terricola, Zygoascus meyerae and Papiliotrema flavescens. Regarding the Sicilian vineyard, a weak association with Hanseniaspora genus or with Leotiomycetes was observed since the results were not confirmed by both analyses (sPLS-DA and LEfSe).
Apart from the well-known role of Hanseniaspora spp. on the quality of some wines (Lombardi et al., 2018; Testa et al., 2020), the exact functions of core taxa fungi or potential biomarker fungi in wine and winemaking are unknown or poorly investigated for most of the taxa identified in the present work, while some more information, even if sometimes in contrast, is available regarding their role on grapevines. For example, some studies reported that Filobasidium spp. was detected on undamaged Tempranillo grapes, but it was not involved in spontaneous must fermentation (Bougreau et al., 2019). Similarly, Aureobasidium would appear to have no impact on the fermentation process and the wine quality (Barata et al., 2012; Setati et al., 2015; Grangeteau et al., 2017). Also, Alternaria spp. is generally not associated with neither fermentation or spoilage of wine (Barata et al., 2012; Setati et al., 2015; Grangeteau et al., 2017), while Cladosporium genus is considered a spoilage of the winery environment (Barata et al., 2012; Grangeteau et al., 2017). Both Cladosporium spp. and Alternaria spp. are endophytic fungi detected in plants showing symptoms of grapevine trunk diseases (GTDs) although their direct role in the disease was not confirmed (Patanita et al., 2022). In other studies, these genera were detected on grapevines, but no visible symptoms of plant diseases were observed (Varanda et al., 2016). Some authors reported that the presence of Cladosporium genus, in particular C. herbarum, on grape berries, is associated with the Cladosporium rot (Barata et al., 2012). In our study, despite the association of C. herbarum with grape samples from Molise vineyard, no disease symptoms were visible on the grapes or grape plants. This could be attributed to the fact that the induction of disease may depend on many factors ascribable to the concurrent presence of this species with other microorganisms or with specific plant factors (Liu and Howell, 2021). Similar contrasting results were also reported regarding Alternaria spp., as some authors reported the potential role of Alternaria spp. as biological control agent against GTDs-associated fungi and other grapevine pathogens (Musetti et al., 2006; Bruez et al., 2014).
Sporobolomyces spp. have been detected in grape musts as well as at the beginning of wine fermentation (Li et al., 2018). These yeasts are not involved in the alcoholic fermentation, but they could contribute to the production of some metabolites that affect the aroma of wines. For instance, it is known that Sporobolomyces roseus can produce volatile compounds such as higher alcohols, acetate esters and thiols (Verginer et al., 2010). Moreover, there are evidence of its potential role as post-harvest biocontrol agent against Penicillium expansum (Sanzani et al., 2021).
5 ConclusionMicrobial terroir involves multiple interacting aspects, such as soil and plant-associated microbes and plant-microbe interactions. The present metataxonomic study explored the distribution of fungal communities at vineyard scale considering different vineyards located in Southern Italian regions dedicated to the production of Aglianico and Cabernet wines. Our results, although worthy of further investigation, highlighted the relevance of the vineyard location in the definition of grape-associated fungal communities. Moreover, from the results obtained in the present study, it appeared that the fungal community present on grape berries is more associated with vineyard location than with the grapevine cultivar, allowing to consider some fungal groups as territorial biomarkers regardless of the cultivar analyzed. For instance, in our study, the Phaeosphaeriaceae family was associated to the Campanian vineyard, while Cladosporium herbarum, Sporobolomyces roseus, and Alternaria metachromatica resulted significantly correlated to samples from Molisian vineyard and Leotiomycetes to the Sicilian one, and this for both varieties analyzed. Of course, further studies are needed, as numerous variables such as growing season, soil properties, plant status and other factors may contribute to the definition of fungal communities typical of vineyards located in near geographical areas.
Data availability statementThe datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://data.mendeley.com/datasets/tp2rrdz8wp/2, doi: 10.17632/tp2rrdz8wp. https://data.mendeley.com/drafts/6jcyrwj2gh, doi: 10.17632/6jcyrwj2gh.
Author contributionsMI: Conceptualization, Validation, Writing – original draft, Writing – review and editing. DB: Formal analysis, Writing – review and editing. FV: Formal analysis, Writing – review and editing. BT: Formal analysis, Writing – review and editing. PT: Conceptualization, Writing – review and editing. MS: Conceptualization, Writing – original draft, Writing – review and editing. GP: Data curation, Software, Writing – original draft, Writing – review and editing. FL: Formal analysis, Writing – review and editing. GA: Formal analysis, Writing – review and editing. SJL: Formal analysis, Writing – review and editing. RC: Conceptualization, Funding acquisition, Supervision, Validation, Writing – review and editing.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was carried out in the context of the scientific project ADAPT: influence of agro-climatic conditions on the microbiome and genetic expression of grapevines for the production of red wines: a multidisciplinary approach. PRIN: Research Projects of National Interest; funded by the Italian Ministry for Universities and Research (MUR), grant number 2017M83XFJ-(CUP H34I19000590001).
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1399968/full#supplementary-material
ReferencesAbarenkov, K., Zirk, A., Piirmann, T., Pöhönen, R., Ivanov, F., Nilsson, R. H., et al. (2022). UNITE QIIME Release for Fungi. Version 16.10.2022. Irvine: UNITE Community. doi: 10.15156/BIO/2483915
PubMed Abstract | Crossref Full Text | Google Scholar
Barata, A., Malfeito-Ferreira, M., and Loureiro, V. (2012). The microbial ecology of wine grape berries. Int. J. Food Microbiol. 3, 243–259. doi: 10.1016/j.ijfoodmicro.2011.11.025
PubMed Abstract | Crossref Full Text | Google Scholar
Belda, I., Zarraonaindia, I., Perisin, M., Palacios, A., and Acedo, A. (2017). Corrigendum: From vineyard soil to wine fermentation: Microbiome approximations to explain the “terroir” concept. Front. Microbiol. 8:1065. doi: 10.3389/fmicb.2017.01065
PubMed Abstract | Crossref Full Text | Google Scholar
Bleidorn, C. (2015). Third generation sequencing: technology and its potential impact on evolutionary biodiversity research. Syst. Biodivers. 14, 1–8. doi: 10.1080/14772000.2015.1099575
Crossref Full Text | Google Scholar
Bodor, A., Bounedjoum, N., Vincze, G. E., Kis, A. E., Laczi, K., Bende, G., et al. (2020). Challenges of unculturable bacteria: environmental perspectives. Rev. Environ. Sci. Biotechnol. 19, 1–22. doi: 10.1007/s11157-020-09522-4
Crossref Full Text | Google Scholar
Bokulich, N. A., Collins, T. S., Masarweh, C., Allen, G., Heymann, H., Ebeler, S. E., et al. (2016). Associations among wine grape microbiome, metabolome, and fermentation behavior suggest microbial contribution to regional wine characteristics. mBio 7:e00631-16. doi: 10.1128/mbio.00631-16
PubMed Abstract | Crossref Full Text | Google Scholar
Bokulich, N. A., Thorngate, J. H., Richardson, P. M., and Mills, D. A. (2014). Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. U. S. A. 111, E139–E148. doi: 10.1073/pnas.1317377110
PubMed Abstract | Crossref Full Text | Google Scholar
Bougreau, M., Ascencio, K., Bugarel, M., Nightingale, K., and Loneragan, G. (2019). Yeast species isolated from Texas High Plains vineyards and dynamics during spontaneous fermentations of Tempranillo grapes. PLoS One 14:e0216246. doi: 10.1371/journal.pone.0216246
PubMed Abstract | Crossref Full Text | Google Scholar
Bruez, E., Vallance, J., Gerbore, J., Lecomte, P., Da Costa, J. P., Guerin-Dubrana, L., et al. (2014). Analyses of the temporal dynamics of fungal communities colonizing the healthy wood tissues of esca leaf-symptomatic and asymptomatic vines. PLoS One 9:e95928. doi: 10.1371/journal.pone.0095928
留言 (0)