
記住我


The TAVI XS trial is an investigator-initiated, multicentre, randomised trial comparing an upper extremity approach to a lower extremity approach regarding secondary access during TAVI. Participating centres are Radboud University Medical Centre (Nijmegen, The Netherlands), Amsterdam University Medical Centre (Amsterdam, The Netherlands), Catharina Hospital (Eindhoven, The Netherlands) and St. Antonius Hospital (Nieuwegein, The Netherlands). This trial has been approved by the Medical Research Ethics Committee Oost-Nederland and by the institutional review board of each participating site. Written informed consent is obtained from all patients prior to enrolment. The trial was designed in accordance with the Declaration of Helsinki. All data are collected in Castor (Castor EDC, Amsterdam, The Netherlands). Evaluation of serious adverse events (SAEs) is performed by an independent data safety monitoring board (DSMB), which convenes when 50% of the patients have reached the 30-day follow-up. A clinical event committee will review and adjudicate all endpoint-related AEs. Monitoring of the trial is executed by the Radboudumc Technology Centre Clinical Studies. The TAVI XS trial has been registered in the ClinicalTrials.gov database (NCT05672823).
InclusionAll patients ≥ 18 years scheduled for transfemoral TAVI are screened for inclusion. Patients with a contra-indication for upper arm or femoral vein access, contra-indication for radial or femoral artery access, or patients in whom there is an intent to use a cerebral embolic protection device requiring additional (arterial) access are excluded (Fig. 1).
Fig. 1
Flowchart of the TAVI XS trial. BARC Bleeding Academic Research Consortium, QuickDASH Quick Disabilities of Arm, Shoulder and Hand, LEFS Lower Extremity Functional Scale, TAVI transcatheter aortic valve implantation, VARC Valve Academic Research Consortium
RandomisationEligible patients are randomly assigned to receive one of the two study treatments in a 1:1 ratio. Randomisation is performed within Castor, with variable block sizes of 2 and 4, and stratification according to study site and use of dual antiplatelet therapy and/or oral anticoagulants at baseline.
Study endpointsThe primary endpoint is defined as clinically relevant bleeding (i.e. Bleeding Academic Research Consortium (BARC) type 2, 3 or 5) [9] of the randomised secondary access site (either diagnostic or pacemaker access, or both) within 30 days after TAVI. If clinically relevant bleeding occurs at both the diagnostic and pacemaker access site, the highest classification of the two BARC bleedings is scored. Secondary endpoints include time to mobilisation, duration of hospitalisation, any BARC type 2, 3 or 5 bleeding, extremity dysfunction score (assessed with the Quick Disabilities of the Arm, Shoulder and Hand [10] and Lower Extremity Functional Scale [11] questionnaires at baseline and 1‑month follow-up) and early safety (30 days) as defined by Valve Academic Research Consortium‑3 criteria [12].
Efficacy endpoints include cross-over rate to the non-randomised secondary access site (defined as conversion from upper to lower extremity or vice versa, which applies to either diagnostic or pacemaker access, or both; conversion to the contralateral upper or lower extremity is not considered a cross-over, but these data will be collected), number of punctures of the randomised secondary access site, fluoroscopy time, procedural time and incidence of temporary pacemaker failure.
TAVI procedureTAVI is performed according to the local protocol of each participating site. In patients randomised to the upper extremity approach, the radial artery is used for diagnostic access. An upper arm vein is used for temporary pacemaker access (Fig. 2); alternatively, pacing over the left ventricular (LV) stiff wire can be used (left to the operator’s discretion). In patients randomised to the lower extremity approach, the contralateral femoral artery (femoral artery not used for TAVI access) is used for diagnostic access. The femoral vein is used for temporary pacemaker access; alternatively, pacing over the wire can be used (operator’s discretion). The decision regarding venous access or pacing over the wire is made prior to randomisation. When pacing over the wire is performed, venous access will not be routinely obtained. All sites are instructed to use ultrasound (US) guidance to obtain femoral artery, femoral vein and upper arm vein access. The radial artery could also be punctured using US guidance, but this is left to the discretion of the operator.
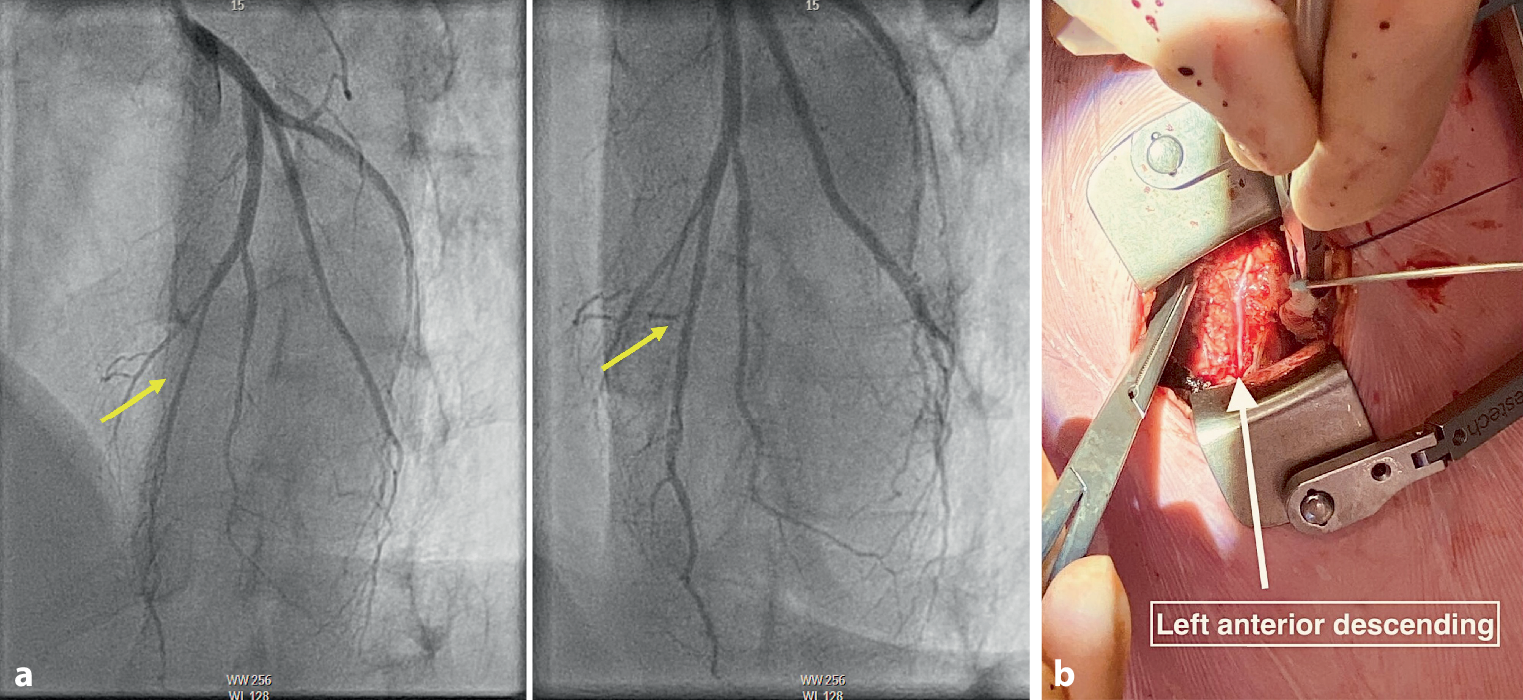
Fig. 2
a–g Procedural steps in temporary pacing lead placement in an upper arm vein. a Positioning of the upper arm. b, c Ultrasound-guided visualisation of the upper arm vein. Arrow indicates the upper arm vein. d Guidewire entering the upper arm vein. Arrow indicates the guidewire. e 6F sheath inserted in the upper arm vein. f Fixation of 5F temporary pacing lead inserted through a 6F sheath in the upper arm vein. g Angiographic visualisation of the temporary pacing lead trajectory from a left upper arm vein to right ventricular apex
Upper arm venous access is obtained by puncturing a robust vein without relevant anatomical structures in its direct proximity. A tourniquet is used to improve visualisation of the vein. A more detailed instruction of this approach has been described previously [13].
Follow-upFollow-up is performed at discharge and at 30 days post-TAVI, either on-site or by phone call. All SAEs are documented from inclusion to 30-day follow-up and are assessed by an independent DSMB, composed of two experienced cardiologists and an epidemiologist/statistician.
Sample size calculation and statisticsWe anticipate an incidence of the primary endpoint of 2% in the upper extremity group and 12% in the lower extremity group, based on previous studies investigating the bleeding rate of secondary access sites [8, 14]. Based on a superiority design with a type I error of 5% and a power of 80%, a total of 216 patients will be needed. Assuming a 10% loss to follow-up rate, a total of 238 patients will be needed.
The primary analysis will take place after the last patient follow-up, using an intention-to-treat approach. Categorical data will be presented as frequencies and proportions. Continuous data will be presented as mean and standard deviation or as median and interquartile range, as appropriate. The chi-square test will be used for the primary endpoint. Superiority will be tested to evaluate the safety of an upper extremity over a lower extremity approach. Superiority is proven if the two-sided p-value is < 0.05. Besides the intention-to-treat analysis, a secondary, separate ‘as treated’ analysis will be performed for the primary endpoint.
留言 (0)