Cell lines and reagents
Human LUAD cell lines (H1792, H2009, H23, A549, H2030, H1437, H1568, and H2126) and murine LUAD cell line Lewis Lung Carcinoma (LLC) were purchased from the American Type Culture Collection. KLA LUAD cells were derived from KR181fl/fl mice [25]. 393P cells were derived from KRASLA1/+; p53R172HDG mice, and provided by Dr. J. M. Kurie (The University of Texas, MD Anderson Cancer Center, Houston, TX, USA). Lacun3 LUAD cell line, which was established from a chemically induced LUAD mice model [26], was a gift of Prof. L. Montuenga (Cima Universidad de Navarra, Pamplona, Spain). CMT167 LUAD cell line was kindly provided by Dr. F. Torres Andon (Center for Research in Molecular Medicine and Chronic Diseases, Santiago de Compostela, Spain). Murine and human cells were cultured at 37 °C and 5% CO2 in DMEM (Gibco, Waltham, MA, USA) or RPMI 1640 medium (Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum and 1% Penicillin/Streptomycin (Gibco, Waltham, MA, USA). Cells were routinely tested for Mycoplasma with the MycoAlert Mycoplasma Detection Kit (Lonza, Basel, Switzerland).
Trametinib, GSK2126458 and proteasome inhibitor (MG-132) #S2619 were acquired from Selleck chemicals (Houston, TX, USA). Trametinib was resuspended in dimethyl sulfoxide (DMSO) (Sigma-Aldrich, MO, USA) for in vitro experiments and in corn oil (Sigma-Aldrich, MO, USA) for in vivo assays. Hydroxychloroquine sulfate H0915 (CQ) MFCD00078203 was obtained from Sigma-Aldrich (MO, USA).
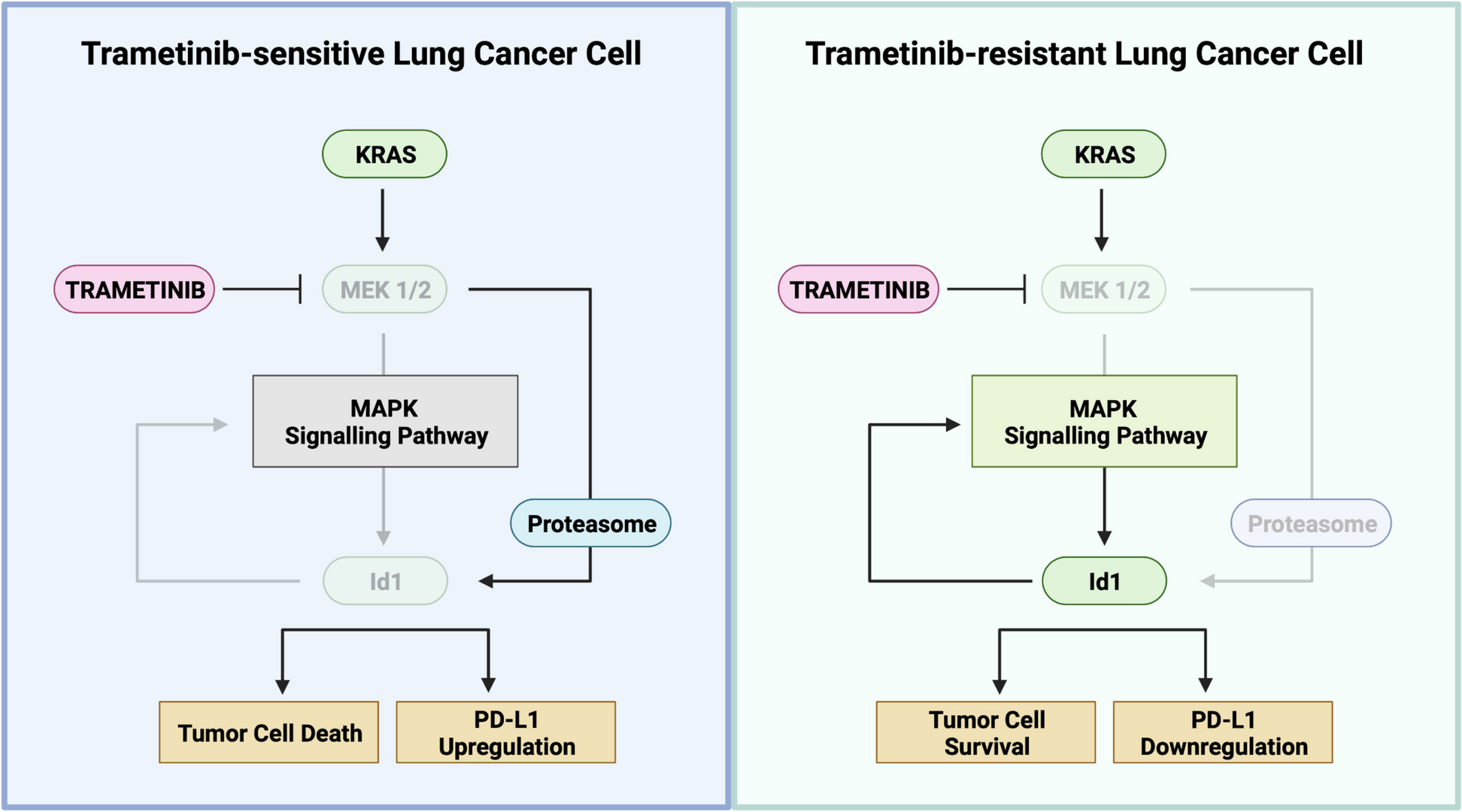
Trametinib-resistant (TR) cell lines were generated by culturing murine (CMT167, LLC and KLA) and human (H1792, H2009, A549 and H2030) parental lines with increasing concentrations of trametinib up to 500 nM, as previously described [27]. TR cell lines were always cultured in cell media with trametinib (500 nM) which was renewed every 4 days. TR-cell resistance to trametinib was measured by analyzing the proliferation capacity of these cells in the presence of trametinib, and compared with parental cells.
In vitro assays
Id1 silencing was performed as previously described [19, 23, 24]. The expression of Id1 was upregulated by lentiviral transduction with mouse or human Id1-flag cDNA expressing vectors (Id1-flag) or retroviral transduction of human Id1 cDNA (Id1-OE) as previously described [19, 24]. Transduction with a GFP cDNA expressing vector was used as control. Oligonucleotides for SMURF2 shRNA (TRCN0000027749) were: Forward sequence: 5'-CCGGCCACACTTGCTTCAATCGAATCTCGAGATTCGATTGAAGCAAGTGTGGTTTTTG-3' Reverse sequence: 5'-AATTCAAAAACCACACTTGCTTCAATCGAATCTCGAGATTCGATTGAAGCAAGTGTGG-3' (Sigma-Aldrich, MO, USA). Oligonucleotides were annealed and cloned into the lentiviral plasmid pLKO (Addgene plasmid #21,915) (MA, USA).
Western blot analysis were performed as previously described [23]. The antibodies used and their dilutions are indicated in Supplementary Table S1. The quantification of the Western blot band intensity was done using Image Lab software (Bio-Rad, CA, USA), and normalized with the density of the loading control protein. RNA extraction and real-time PCR were performed as previously described [23]. The sequences of the primers used in the real-time PCR assays are indicated in Supplementary Table S2.
For proliferation assays, 2500 cells per well were seeded in 96-well plates in 100 μL of complete medium in triplicate for each experimental condition. Cell proliferation was assessed at 72 h using trametinib concentrations ranging from 2000 nM to 0.008 nM. Cells were fixed with formaldehyde and stained with a commercial 1% crystal violet solution (V5265, Sigma-Aldrich, Saint Louis, MO, USA). A 20% acetic solution was used to dissolve crystal violet, and the absorbance was measured at 590 nm in a SPECTROstar Nano (BGM LABTECH, Ortenberg, Germany).
RNA sequencing
RNA sequencing data analysis was performed using the following workflow: (1) the quality of the raw reads was verified using FastQC software and the trimming of the reads was carried out with trimmomatic [28]; (2) alignment against the mouse reference genome (GRCh38) was performed using STAR [29]; (3) gene expression quantification using read counts of exonic gene regions was carried out with featureCounts [30]; (4) the gene annotation reference was Gencode v42 [31]; and (5) differential expression statistical analysis was performed using R/Bioconductor (https://www.R-project.org/) as follows. Gene expression data was normalized with edgeR [32] and voom [33]. After quality assessment and outlier detection using R/Bioconductor, a filtering process was performed. Genes with read counts lower than 4 in more than the 50% of the samples of all the studied conditions were considered as not expressed. LIMMA was used to identify the genes with significant differential expression between experimental groups. Genes were selected as differentially expressed using a cut-off value of B > 5. Further functional and clustering analyses and graphical representations were performed using the R/Bioconductor and clusterProfiler [34]. Data are publicly available in GEO database with the accession number GSE236258.
Mouse lung cancer models and therapeutic schedules
All animal procedures were approved by the institutional Committee on Animal Research and Ethics (regional Government of Navarra) under the protocol numbers E17-22(054-19E2), E18-21(054-19E2) and E24-20(049-E1). This study included 8–12 week-old female C57BL/6 J mice (Envigo, Indianapolis, IN, USA) and 8 week-old Sv/129 female mice (Janvier Labs, Le Genest-Saint-Isle, France).
LLC cells (1.5 × 106), LLC cells expressing Id1-flag (1.5 × 106) or CMT167 cells (2 × 105) were subcutaneously inoculated in the right flank of syngeneic mice. LLC and CMT167 engraftments were allowed to grow for 7 days, and then treated with vehicle or with trametinib (1 mg/kg; oral administration, 5 times a week) until day 30.
To assess the antitumor effect of trametinib and its combination anti-PD-1, LLC or 393P (4 × 106) cells were subcutaneously injected in the right flank of female C57BL/6 J and Sv/129 mice, respectively. LLC and 393P tumors were allowed to grow for 7 and 14 days before treatment, respectively. Tumor-bearing mice were randomized in four groups and treated with anti-PD-1 (100 mg/mouse; intraperitoneally, twice a week; RMP1-14 BioXCell, Lebanon, NH, USA), trametinib (1 mg/kg; oral oral administration, 5 times a week), their combination, or vehicle for three weeks.
Tumors were measured periodically using a digital caliper (DIN862, Ref 112-G, SESA Tools, Hernani, Spain), and tumor volume was calculated using the following formula:
$$\mathrm\;\mathrm\:=\:\mathrm\pi/6\:\;\times\;\:\mathrm\;\;\times\;\mathrm^2$$
Immunohistochemistry (IHC)
For histological analyses, tumors were harvested, fixed in formaldehyde 4% pH = 7 (Panreac, Castellar del Valles, Spain) for 48 h, embedded in paraffin, and sectioned for hematoxylin–eosin staining (H&E). Immunohistochemistry (IHC) for the detection of Id1, CD8, CD3, CD4 and FOXP3 was performed as previously described [19, 24]. Slides were scanned with the Aperio Digital Scanner (Leyca, Wetzlar, Germany) and analyzed with ImageJ (NIH, Bethesda, MD, USA).
Flow cytometry analyses
Tumors were dissected, cut into small pieces, and incubated with 1 mg/ml collagenase D and 50 µg/ml DNase I (Roche, Basel, Switzerland) at 37 °C for 30 min. EDTA (6 µM) was then used to block collagenase and DNase I activities. Then, tumors were mechanically disaggregated through a 70 μm cell strainer (Corning, NY, USA). Erythrocytes were lysed with ACK buffer (Gibco, Waltham, MA, USA). Single cell suspensions were layered over 35% Percoll (GE Healthcare, IL, USA) and centrifuged to purify the tumor infiltrating leukocytes for flow cytometry analysis. Cell suspensions were incubated with a purified monoclonal antibody against CD16/CD32 (Mouse BD Fc Block, 1:200, BD Bioscience, NJ, USA) for 15 min at 4ºC and stained with fluorochrome-conjugated antibodies diluted in FACS buffer (PBS, 5% Fetalclone, 2.5 mM EDTA). For intracellular staining, cells were permeabilized with eBioscience Fixation/Permeabilization Kit (Invitrogen, Waltham, MA, USA) for 15 min and stained. The antibodies used for flow cytometry analyses are listed in Supplementary Table S3. Dead cells were excluded using PromoFluor-840 NIR Maleimide (PromoCell, Heidelberg, Germany). Gating strategies are shown in Supplementary Figs. S1 and S2.
Quantification of apoptosis was assessed by flow cytometry as previously described [19]. PD-L1 expression was assessed by flow cytometry, as previously described [2]. Briefly, LUAD cells were seeded into 6-well plates. After 24 h, human and murine lung cancer cells were treated with trametinib for 72 h. Trametinib concentrations used were 100 nM for murine and 500 nM for human LUAD cells. Control cells were treated with DMSO. After 48 h of incubation, recombinant murine (500 U/mL, PeproTech, MA, USA) or human (20 ng/ml, Biolegend,CA, USA) IFN-γ was added, and the cells were incubated for 24 h. Cells without IFN-γ were also analyzed. PE-conjugated anti-CD274 (PD-L1) was used to detect PD-L1 expression on lung cancer cell surface. Cells were acquired using a CytoFLEX LX flow cytometer (Beckman Coulter, CA, USA) and analyzed with CytExpert software (Beckman Coulter, CA, USA).
Statistical analysis
Data normality was assessed using the Shapiro–Wilk test. Parametric comparations between experimental groups were performed by two-sided t-test or one-way ANOVA test followed by the Tukey post hoc test. Non-parametric comparisons between experimental groups were performed by two-sided Mann–Whitney U-test or Kruskal–Wallis test with post hoc Mann–Whitney U-test. Survival curves were generated using the Kaplan–Meier method, and differences were analyzed with the log-rank test. p < 0.05 was considered statistically significant. Statistical analyses were performed using the version 9 of GraphPad Prism software (GraphPad Prism 9, San Diego, CA, USA).





留言 (0)