In this report, we report 26 new families with distal Xq28 duplication syndrome. Notably, many of our cases were diagnosed incidentally, during pregnancy, even when there was no initial indication for fetal genetic testing. Among the prenatal cases where there was a specific medical indication for CMA testing, such as cases involving VSD, ARSA, and ventriculomegaly, we did not observe any other structural anomalies. It is noteworthy that all these US findings were identified in female fetuses.
While 3 of the 19 prenatal cases within our cohort underwent testing following the detection of fetal sonography findings, including VSD, ARSA, and fetal brain ventriculomegaly, no prenatal phenotype had been previously documented in the 3 prenatal cases published to date. However, according to the 32 postnatal cases previously published, certain congenital anomalies mentioned could theoretically have been diagnosed through sonographic scans during pregnancy, including but not limited to polydactyly, hypospadias, imperforate anus, and others [9].
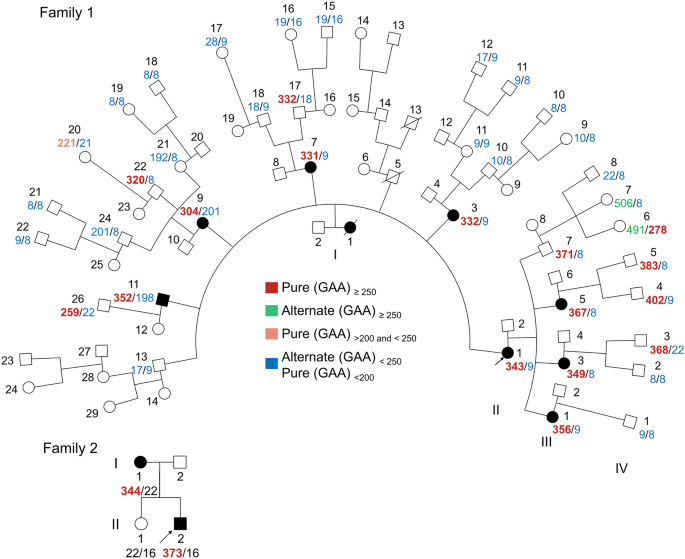
Knowledge about the clinical spectrum of this duplication in adult life is limited. In this study, we present data from 14 family members, primarily parents of the index cases. Notably, within our cohort, we describe four adult males (three transmitting fathers and one maternal uncle) with normal to mild presentations, such as mild ID and ADHD.
In our cohort, a partial allelic duplication of ~200–300 Kb was identified in eight cases. Intriguingly, five of these cases had ancestral ties to the Armenian/Caucasian Jewish population, suggesting the possibility of a founder variant. Further research and genetic analysis are warranted to explore the origins and implications of this potential founder variant in greater detail. However, it is essential to note that since both the “full” and “partial” distal Xq28 duplications encompass the RAB39B and CLIC2 genes, which have been proposed as key contributors to the disease mechanism, genetic counseling remains consistent for both types of duplications.
In our cohort, we did not identify any instances of de-novo duplications. Nevertheless, in the literature, although the majority of affected individuals inherited the duplication from their heterozygous mothers, two cases of de-novo duplications have also been documented [9].
Among the prenatal cases in our cohort, couples chose pregnancy termination in nine instances. Remarkably, two of these cases involved female fetuses with no concurrent pathogenic CNVs. In six cases with female fetuses, pregnancies continued, but no developmental or cognitive follow-up data are currently available. The ethical dilemma surrounding the decision to terminate pregnancies with female fetuses diagnosed with the distal Xq28 duplication is a topic of significant interest. While El-Hattab et al. present six females exhibited a milder phenotype with mild cognitive impairment in the form of learning difficulties, ADHD, and some distinctive facial features [5], Ballout et al. report includes four females, three of whom exhibit neurocognitive impairment [9]. Collectively, Ballout et al. described nine individuals diagnosed with the syndrome, with prenatal diagnoses provided for three cases – one male fetus whose pregnancy was terminated, one female fetus, and one male fetus delivered.
In a similar context, the question arises about whether the duplication justifies PGD. Although, in some cases, the CNV is a founder variant, it remains disease-causing. Notably, only a small subset of three families from our cohort chose to undergo PGD, highlighting the intricate decision-making process involved in reproductive choices.
Given the complexities surrounding the observed duplications, the inclusion of familial data becomes paramount. Among cases where the mode of inheritance was available, nine were maternally inherited, while three were paternally inherited. Notably, all inheriting mothers appeared to be unaffected by the duplication. Three inheriting fathers were identified, with two exhibiting ADHD and the other reported as asymptomatic.
In a relevant study by Leffler et al., which examined 25 reported cases of K/L-mediated Xq28 duplication syndrome, a condition within the same genetic region but involving different genes, males displayed varying degrees of neurocognitive features. Interestingly, one male from a family with this duplication exhibited normal intelligence, suggesting that this genetic variant may not exhibit full penetrance [10]. These observations underscore the importance of family segregation studies, which offer a valuable opportunity to investigate potential modifiers, epigenetic factors, and other genetic variants that could influence the clinical phenotype.
Some notable limitations of our study include the absence of postnatal follow-up data for pregnancies that proceeded to term. This hinders our ability to comprehensively understand the long-term outcomes associated with pregnancies in which the duplication persisted postnatally. Regarding the “normal” cognitive evaluations in adult family members, our assessment is grounded in the clinical geneticists’ observations during genetic counseling, rather than formal quantitative evaluations by cognitive neurologists. Carrier parents, even when influenced by the duplication, often maintain a high level of functionality, allowing them to marry, have children, and undergo prenatal testing. While we are not neurologists or psychiatrists specializing in quantitative IQ assessments, our experience as genetic physicians frequently involve carrier parents, whose self-perceived “non-affected” status is typically sufficient to rule out moderate cognitive impairment and beyond.
Additionally, among the nine cases of Ashkenazi Jewish ancestry with the detected duplication, it is plausible that a founder effect may contribute to its prevalence. However, it remains uncertain whether the prevalence is solely linked to ancestry or attributed to the relatively high frequency of Ashkenazi Jewish population within our local database.
Among one center that participated in the study, 8447 individuals were screened with CMA between 2016 and 2023, leading to the identification of nine affected probands (postnatal). The estimated prevalence is ~1%. The prevalence in the prenatal setting is 0.0017% (16 out of 93,646). Due to shared families across different centers and extended families represented in all participating centers, it is challenging to accurately determine the precise prevalence. Furthermore, assessing penetrance based on a limited number of cases in both male and female individuals proves challenging. Nonetheless, it is prudent to consider that hereditary factors, familial expression, and ancestry may play crucial roles in clinical decision-making regarding pregnancy outcome and future family planning [11, 12].
We did not investigate the precise genomic positioning of the duplication, i.e., whether it occurs in a tandem arrangement or not. It is conceivable that certain variations in clinical phenotype may not be directly attributable to the duplicated genetic content itself, but rather to the specific genomic location or orientation of the duplication on the X chromosome, potentially affecting spatial structural elements [11, 13].

留言 (0)