
記住我

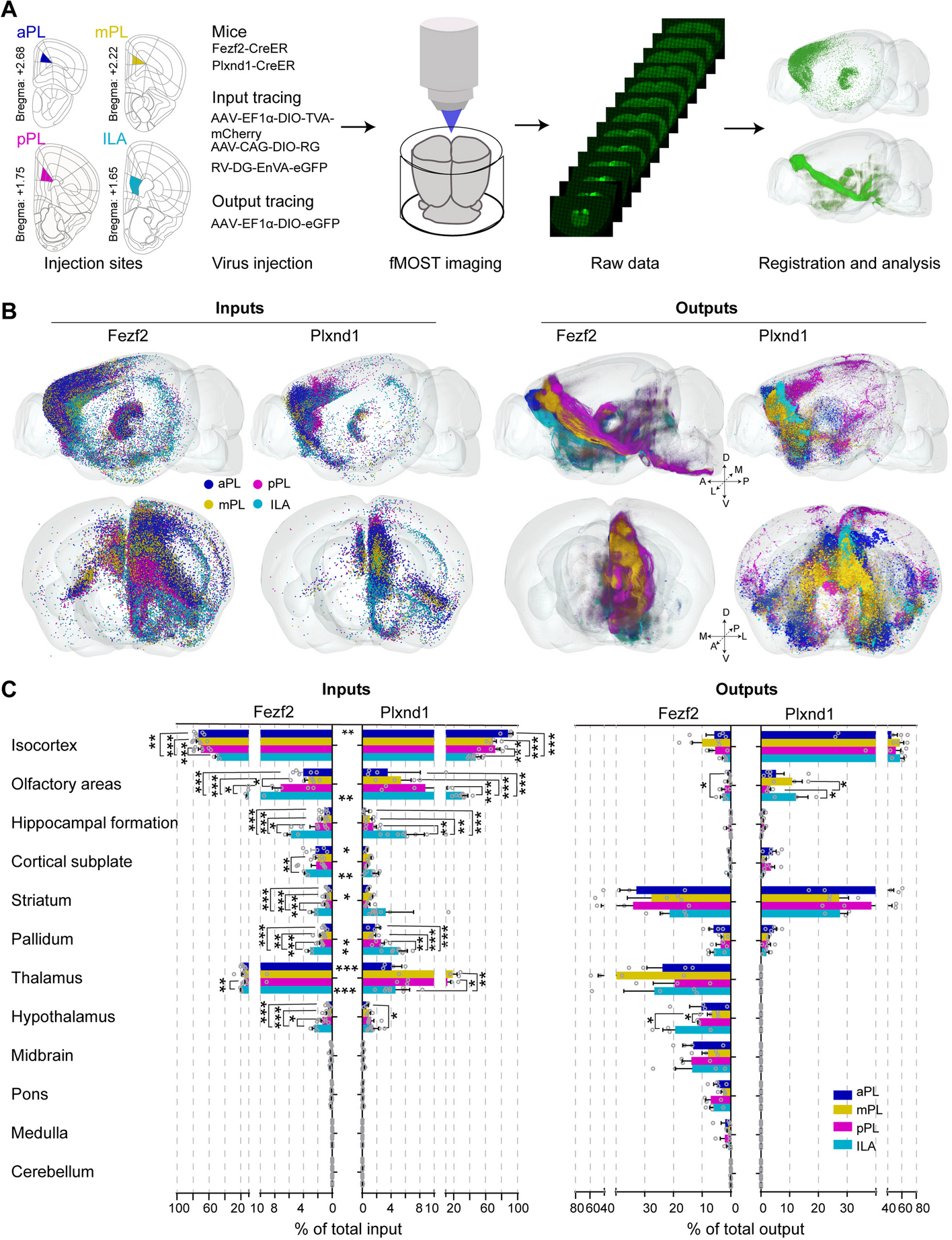
Inspired by our finding that GATA3 functions downstream of BRCA1 to suppress aberrant luminal to basal and mesenchymal differentiation in the induction of BLBCs [31, 32], and the discovery that inadequate DNA damage repair promotes aberrant mammary epithelial cell differentiation [3], we hypothesized that GATA3, like BRCA1, also functions in DNA repair during mammary tumorigenesis. To test this hypothesis, we performed IHC analysis with the antibody against γH2AX, a marker for DNA DSBs, in mammary tumors spontaneously developed in p18−/− and p18−/−;Gata3+/− mice. We found that γH2AX-positive cells were barely detectable in p18−/− tumors; however, significantly more γH2AX-positive cells were found in p18−/−;Gata3+/− tumors (Fig. 1a, b). In line with our previous finding, we confirmed that p18−/− mammary tumors were well-differentiated luminal type and that p18−/−;Gata3+/− tumors were poorly differentiated basal-like with EMT features (Fig. 1c, and details in [31, 32]). These results indicate that deficiency of Gata3 promotes DNA damage accumulation and induces basal and mesenchymal differentiation in mammary tumorigenesis. As a direct comparison, we also performed a similar analysis for mammary tumors spontaneously developed in p18−/−;Brca1+/− mice. As expected, more γH2AX-positive cells were observed in p18−/−;Brca1+/− tumors than in p18−/− tumors (Fig. 1a, b). Notably, the number of γH2AX-positive cells in p18−/−;Gata3+/− tumors was less than that in p18−/−;Brca1+/− tumors, suggesting that the efficiency of DNA damage repair by GATA3 is not as strong as that by BRCA1. We determined tumor cell proliferation in vivo and observed that the percentages of Ki-67-positive cells were comparable in p18−/− and p18−/−; Brca1+/− or p18−/−; Gata3+/− tumors (Fig. 1d, Additional file 1: Fig. S1). These results are consistent with our previous discovery that loss of p18 rescues Brca1- or Gata3-deficient mammary cell proliferation [31, 35], and also suggest that GATA3 deficiency promoted γH2AX-focus formation in vivo is less likely due to cell cycle arrest. Together, these results indicate that a deficiency of Gata3, like that of Brca1, impairs DNA damage repair and promotes aberrant basal and mesenchymal differentiation in mammary tumorigenesis and progression.
Fig. 1
Gata3- and Brca1-deficient mammary tumors display significantly enhanced number of γH2AX-positive tumor cells, and the remaining wild-type Gata3 allele is retained in Gata3 heterozygous tumors. a Representative IHC analysis of mammary tumors spontaneously developed in p18−/−;Gata3+/−, p18−/−;Brca1+/−, and p18−/− mice. Typical γH2AX-positive tumor cells are indicated. The inset shows the enlarged γH2AX-positive cells. b The H-scores for γH2AX in a were calculated. The results represent the mean ± SD of five individual tumors per group. The asterisk (*) denotes a statistical significance from p18−/− and p18−/−;Gata3+/− or p18−/−;Brca1+/− samples determined by the T-test. c Representative mammary tumors were immunostained with antibodies against Ck5 and Ck8. d Analysis of Ki-67-positive cells in mammary tumors. Mammary tumors developed in p18−/−;Gata3+/−, p18−/−;Brca1+/−, and p18−/− mice were analyzed by IHC with an antibody against Ki-67. The percentages of Ki-67-positive cells were calculated. The results represent the mean ± SD of three individual tumors per group. e A representative mammary tumor from p18−/−;Gata3+/− mouse was immunostained with GATA3. Note the heterogeneous GATA3 staining in the tumor cells. The inset shows staining of a normal-like gland in the same mouse. f Presence of the wild-type Gata3 allele in mammary tumors of p18−/−;Gata3+/− mice. DNA extracted from the dissected tumor samples of mice was amplified by PCR to detect wild-type (wt) and mutant (mt) alleles of Gata3. DNA from three p18−/−;Gata3+/− mammary tumors were analyzed and representative results from two tumors were shown
We then performed IHC for p18−/−;Gata3+/− mammary tumors and found that the expression of GATA3 was highly heterogeneous, some of the cells expressed GATA3, and some did not (Fig. 1e). We analyzed genomic DNA isolated from tumors and observed that the remaining wild-type Gata3 allele was retained in all three p18−/−;Gata3+/− mammary tumors tested (Fig. 1f). Given that the remaining wild-type Brca1 allele was lost in CK5-positive basal-like cells, not in CK5-negative cells, in Brca1 heterozygous mammary tumors [35], and that GATA3 functions downstream of BRCA1 to suppress breast cancer [32], it will be interesting to determine if LOH of Gata3 occurs in certain groups of Gata3 heterozygous mammary tumor cells.
Depletion of Gata3 in luminal tumor cells promotes DNA damage accumulation and produces BLBCsTaking advantage of the luminal type mammary tumor model system we established [31, 32], we directly test if Gata3 deficiency impairs DNA damage repair in luminal tumor cells in vivo. We knocked down Gata3 in MMTV-PyMT tumor cells that were isolated and screened from MMTV-PyMT mammary tumors and were confirmed as Gata3 and Brca1 proficient (Gata3+/+;Brca1+/+) luminal type before and after transplantation into mammary fat pads (MFPs) of recipient mice (Fig. 2a, and details in [31, 32]). We transplanted MMTV-PyMT luminal tumor cells into MFPs of mice and performed IHC analysis for newly generated mammary tumors. We found that the tumors generated by Gata3-depleted cells displayed significantly more γH2AX-positive cells than tumors generated by control cells (Fig. 2b, c, Additional file 1: Fig. S2). Together with our previous finding that depletion of Gata3 in these luminal tumor cells promotes basal-like differentiation in tumors newly generated [31], these data demonstrate that depletion of Gata3 in luminal tumor cells also impairs DNA damage repair and promotes basal-like differentiation.
Fig. 2
Depletion of Gata3 in MMTV-PyMT luminal tumor cells enhances DNA damage in tumorigenesis. a Luminal mammary tumor cells from MMTV-PyMT mice were infected with psi-LVRU6GP-control (sh-Ctrl) or psi-LVRU6GP-Gata3 targeting different sequences of mouse Gata3 (sh-Gata3-a and sh-Gata3-c), selected with puromycin, and analyzed for Gata3 expression. b MMTV-PyMT luminal tumor cells infected with sh-Ctrl and sh-Gata3-c were transplanted into the mammary fat pads (MFPs) of female NCG mice. Tumors formed 8 weeks after transplantation were immunostained with an antibody against γ-H2AX. c The H-scores for γ-H2AX in b were calculated. The results represent the mean ± SD of three individual tumors per group. The asterisk (*) denotes a statistical significance from sh-Ctrl and sh-Gata3-c samples determined by the T-test
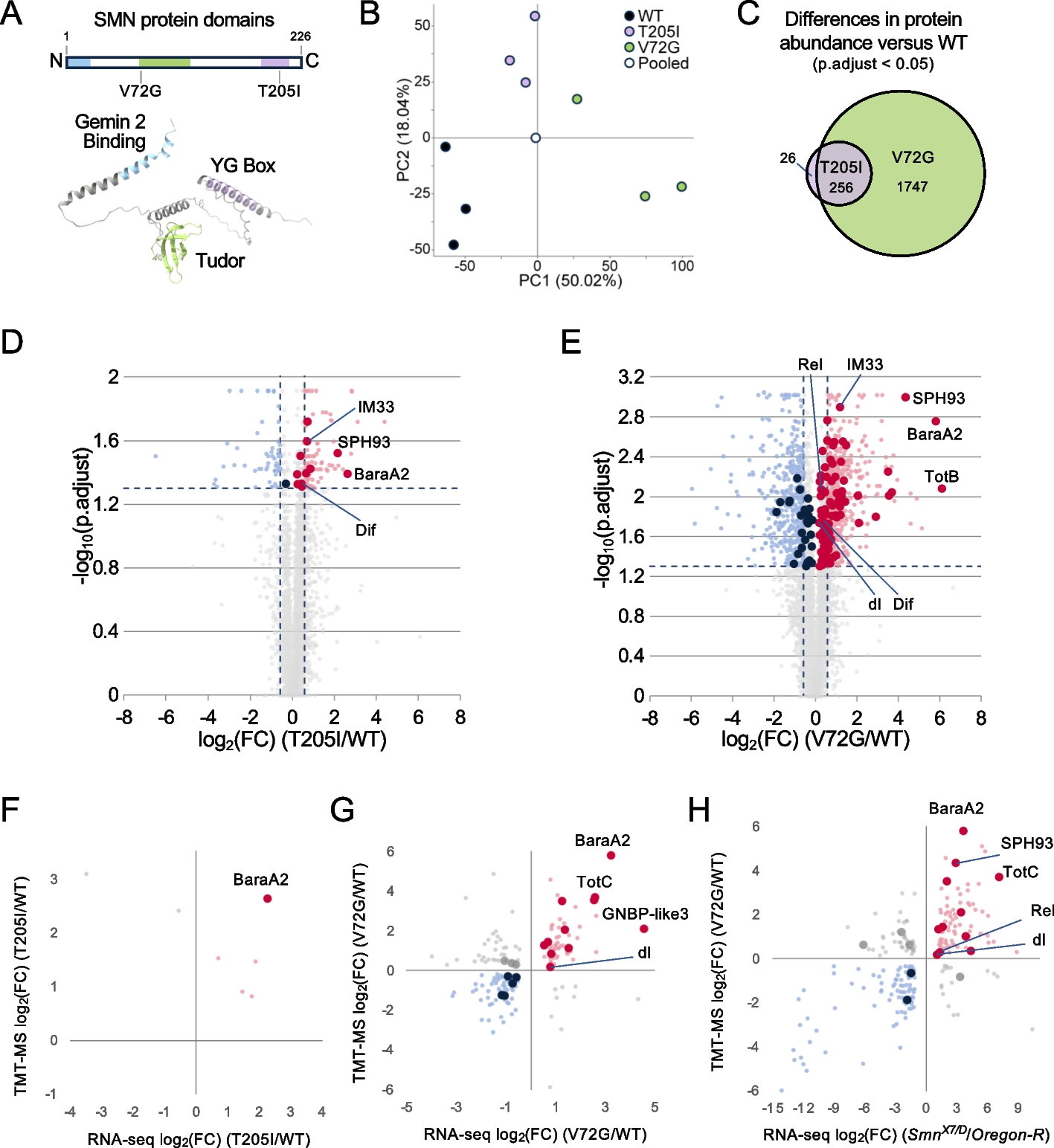
Depletion of GATA3 in breast cancer cells impairs DNA damage response in vitroIt has been reported that depletion of GATA3 in MCF7 cells reduces the expression of CtIP and impairs etoposide-induced DNA damage repair and that the overexpression of WT GATA3, but not the overexpression of a few mutants including D335E, promotes HR [33]. However, since MCF7 cells harbor a heterozygous mutant GATA3 D335E allele and express a barely detectable WT GATA3 protein [22, 32, 36], whether the loss of function of WT GATA3 impacts DNA damage repair remains to be confirmed. We knocked down GATA3 in T47D cells expressing WT GATA3 and harboring a mutant p18 [36, 37] and noticed that depletion of GATA3 downregulated the expression of CtIP and upregulated the expression of γH2AX (Fig. 3a, Additional file 1: Fig. S3). We also checked the expression of RNF-8 and Rad51, both of which play a critical role in DSB repair. We found that the knockdown of GATA3 slightly reduced the expression of RNF-8 and enhanced that of Rad51 (Fig. 3a). We then challenged these cells with etoposide (VP16), a widely used DSB-generating agent (4, 33). We noticed that 20 h after VP16 treatment the percentages of γH2AX- or 53BP1-positive cells were significantly higher in Si-GATA3 cells than in Si-control cells, indicating that depletion of WT GATA3 results in less efficient γH2AX or 53BP1 clearance and that GATA3 deficiency impairs DNA DSB repair (Fig. 3b–d, Additional file 1: Fig. S3, S4). We also determined if the treatment affects the number of T47D cells expressing cyclin A, an S-phase and cell proliferation marker. We observed that the percentages of cyclin A-positive cells were comparable in T47D-Si-GATA3 and T47D-Si-control cells treated with VP16. Notably, after VP16 treatment, some of the γH2AX-focus-positive T47D-Si-GATA3 cells co-expressed cyclin A (Fig. 3b, Additional file 1: Fig. S3b). These results also suggest that the VP16-induced higher levels of γH2AX in Gata3-deficient cells are less likely due to cell cycle arrest. In accordance with the findings derived in GATA3-deficient T47D cells, we also determined the effect of VP16 in the induction of γ-H2AX and 53BP1 in GATA3-deficient U2OS cells and obtained similar results (Additional file 1: Fig. S5).
Fig. 3
Depletion of GATA3 in breast cancer cells impairs DNA damage repair in vitro. a Human luminal breast cancer cell line, T47D, was transfected with Si-GATA3-1, Si-GATA3-2, Si-GATA3-3, or Si-control (Si-Ctrl) and analyzed. b–d T47D cells were transfected with Si-GATA3-1, Si-GATA3-2, or Si-Ctrl for 32 h, treated with VP16 at a concentration of 10 μM for additional 0.5 h, allowed to recover for 0 or 20 h, and then immunostained with an antibody against γH2AX and cyclin A (b, c) or 53BP1 (d). Representative γH2AX- and cyclin A-positive cells were shown. Note relative to T47D-Si-Ctrl cells, T47D-Si-GATA3-1 and T47D-Si-GATA3-2 cells retain high levels of γH2AX after 0- and 20-h recovery. γH2AX and cyclin A doubly positive cells are indicated by white arrows. c Quantification of γH2AX-positive cells in b. Only cells with at least 5 γH2AX foci were counted as positive cells. At least 200 cells were counted for each sample. The results represent the mean ± SD of five randomly selected fields per group. The asterisk (*) denotes a statistical significance from Si-Ctrl and Si-GATA3-1 or Si-GATA3-2 samples determined by the T-test. d Quantification of 53BP1-positive cells. Only cells with at least 3 53BP1 foci were counted as positive cells. At least 200 cells were counted for each sample. The results represent the mean ± SD of five randomly selected fields per group. e Analysis of Gata3 levels in MMTV-PyMT (Gata3+/+;Brca1+/+), p18−/−;Gata3+/− (Gata3+/−), and p18−/−;Brca1+/− (Brca1+/−) mouse mammary tumor cells. f, g Gata3+/+;Brca1+/+, Gata3+/−, and Brca1+/− tumor cells were treated with VP16 (f) for 3 h, or E2 (g) for 24 h. h, i The expression of γ-H2AX in f was quantified in h and that in g was quantified in i. j, k T47D-Sh-Ctrl, T47D-Sh-GATA3-1, and T47D-Sh-GATA3-2 cells were treated with E2 for 1 h, and the expression of genes was determined (j) and quantified (k). The blots, images, and graphs represent data from at least three independent experiments
We then analyzed primary murine mammary tumor cells isolated, screened, and characterized in our previous studies [31, 32]. Since p18−/− luminal tumor cells proliferate very slowly in vitro and weakly generate luminal tumors when transplanted in vivo [32], we utilized a few Gata3+/+;Brca1+/+ luminal tumor cell lines isolated from MMTV-PyMT mammary tumors (Fig. 3e) (this study and reference [31]). We confirmed that Gata3+/+;Brca1+/+ mammary tumor cells expressed high levels of GATA3 and Brca1+/− (p18−/−;Brca1+/−) or Gata3+/− (p18−/−;Gata3+/−) mammary tumor cells expressed very weak GATA3 (Fig. 3e), as we previously described [31, 32]. We found that the treatment of Gata3+/− (p18−/−;Gata3+/−) mouse mammary tumor cells with VP16 led to significantly more γH2AX than the treatment of Gata3+/+;Brca1+/+ (MMTV-PyMT) tumor cells (Fig. 3f, h). As a control, we conducted a similar treatment for Brca1+/− (p18−/−;Brca1+/−) mouse mammary tumor cells and also observed a significant increase of γH2AX in Brca1+/− cells relative to that in Gata3+/+;Brca1+/+ cells (Fig. 3f, h). Interestingly, VP16 did not clearly alter the expression of Rnf-8 and Rad51 (Fig. 3f). These results confirm the role of Gata3 loss, like Brca1 loss, in inducing DNA DSBs in tumor cells.
We then tested whether depletion of GATA3 in mammary tumor cells impairs DNA damage response to estrogen, a hormone fluctuating during the menstrual cycle and inducing DNA DSBs [38]. We found that in response to estrogen (17β-estradiol, E2) Gata3+/− and Brca1+/− mouse mammary tumor cells expressed higher levels of γH2AX than Gata3+/+;Brca1+/+ tumor cells (Fig. 3g, i). Consistently, the expression of CtIP in Gata3+/− tumor cells was lower than in Gata3+/+;Brca1+/+ cells (Fig. 3g). T47D-sh-GATA3-1 and T47D-sh-GATA3-2 cells expressed more γH2AX than T47D-sh-control cells when they were treated with E2 (Fig. 3j, k). These data demonstrate that the depletion of GATA3 or BRCA1 promotes DSB-repairing defects induced by estrogen.
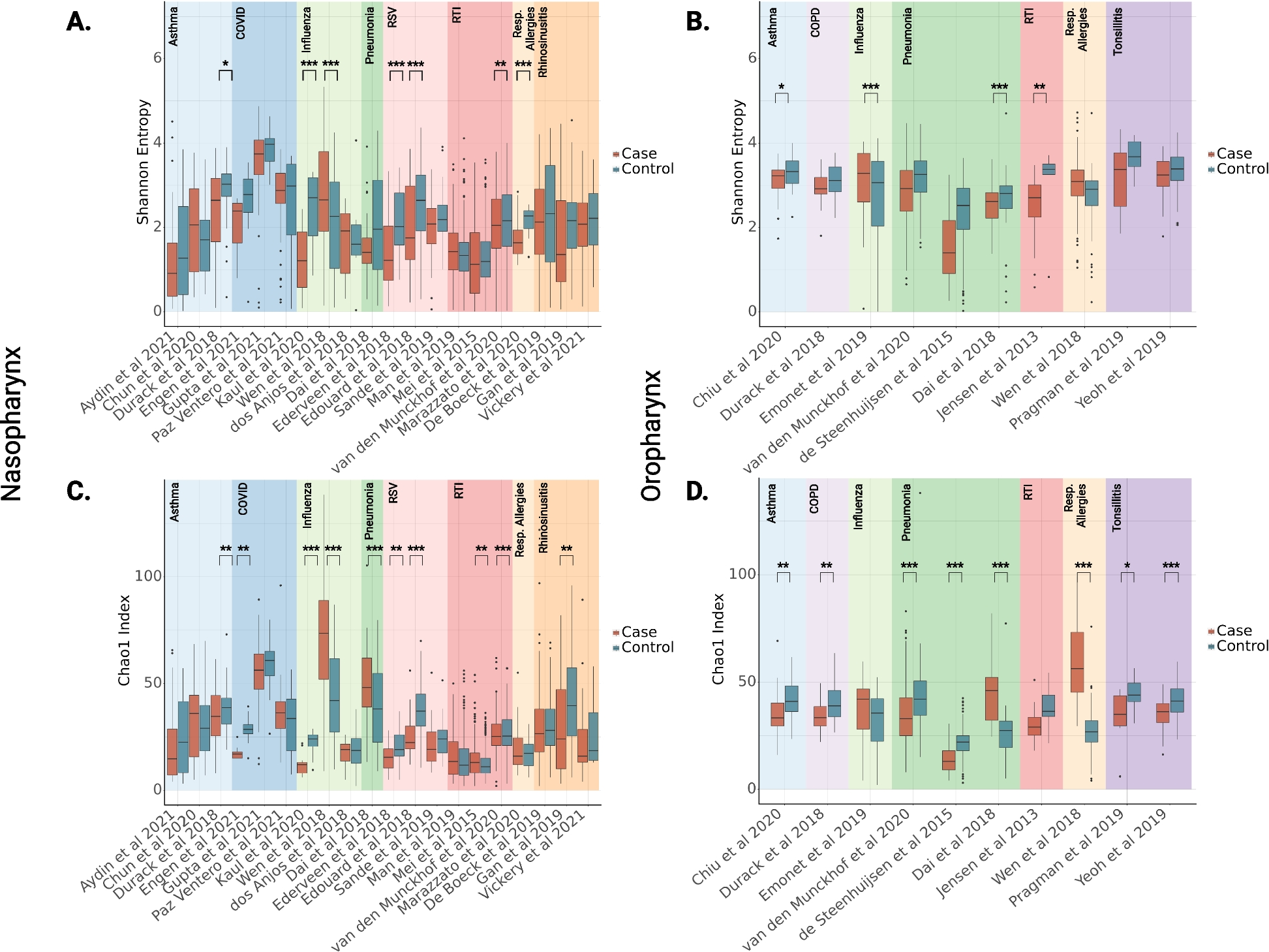
Depletion of Gata3 or Brca1 promotes estrogen-induced DNA damage in mammary tumorigenesisSince estrogen is an intrinsic source of the induction of DNA damage in mammary epithelial and tumor cells under physiological condition, we then transplanted p18−/− (Gata3+/+;Brca1+/+), p18−/−;Gata3+/− (Gata3-deficient), and p18−/−;Brca1+/− (Brca1-deficient) mammary tumor cells into MFPs of the recipient mice who were simultaneously administered estrogen or vehicle. We found that the number of γH2AX-positive cells was significantly more in estrogen-treated p18−/−;Gata3+/− and p18−/−;Brca1+/− tumors than in vehicle-treated counterparts (Fig. 4a–c). Because p18−/− luminal tumor cells did not generate tumors when they were transplanted into MFPs without exogenous estrogen [32], we were unable to determine and compare the effect of estrogen in inducing DNA damage in p18−/− (Gata3+/+;Brca1+/+) luminal type tumorigenesis. Nevertheless, the number of γH2AX-positive cells in estrogen-treated p18−/− tumors was significantly less than the number of γH2AX-positive cells in estrogen-treated p18−/−;Gata3+/− and p18−/−;Brca1+/− tumors (Fig. 4a–c), indicating depletion of Gata3 or Brca1 promotes estrogen-induced DNA damage in tumorigenesis. The finding that Brca1 depletion promotes estrogen-induced DNA damage in vivo is consistent with the previous observation that BRCA1 deficiency exacerbates estrogen-induced DNA damage in vitro [38]. Interestingly, estrogen-treated p18−/−;Gata3+/− tumors exhibited less γH2AX-positive cells than estrogen-treated p18−/−;Brca1+/− tumors. This result is in line with the findings derived from primary p18−/−;Gata3+/− and p18−/−;Brca1+/− mammary tumors (Fig. 1), suggesting Gata3-mediated DNA damage repair may not be as efficient as Brca1-mediated repair in tumor development. Taking into consideration our previous discovery that estrogen promotes activation of EMT in Gata3- or Brca1-deficient tumors accelerating mammary tumorigenesis and metastasis [32, 39], these data demonstrate that deficiency of Gata3, like that of Brca1, promotes estrogen-induced DNA damage and estrogen-activated aberrant differentiation in mammary tumorigenesis and progression.
Fig. 4
Depletion of Gata3 or Brca1 promotes estrogen-induced DNA damage in mammary tumorigenesis. a, b Representative IHC analysis of regenerated p18−/−;Gata3+/−, p18−/−;Brca1+/−, and p18−/− mammary tumors treated with or without E2. Typical γH2AX-positive tumor cells are indicated. c The H-scores for γH2AX in a and b were calculated. The results represent the mean ± SD of four individual tumors per group. The asterisk (*) denotes a statistical significance from E2 and placebo-treated samples determined by the T-test. The asterisk (**) denotes a statistical significance from E2-treated p18−/− and p18−/−;Gata3+/− or p18−/−;Brca1+/− samples determined by the T-test. Note since p18−/− tumor cells did not generate tumors when transplanted without exogenous E2, we calculated γH2AX H scores from primary tumors as an alternative control for placebo-treated p18−/− tumors (indicated by #)
Reconstitution of Gata3 improves DNA damage repair in Brca1-deficient mammary tumor cells and tumorigenesisPrompted by the data that the loss of function of GATA3 generates similar phenotypes with that of BRCA1 with respect to impairing DNA damage repair (Figs. 1, 2, 3, and 4) and that GATA3 functions downstream of BRCA1 to suppress aberrant mammary cell differentiation [31, 32], we then examine whether GATA3 also functions downstream of BRCA1 to control DNA repair. We took advantage of mammary tumor cell lines derived from p18−/−;Brca1MGKO mice, in which both Brca1 and Gata3 were undetectable [12, 32]. We transduced p18−/−;Brca1MGKO mammary tumor cells with pLVX-Flag or pLVX-Flag-Gata3 or transfected human MDA-MB231 cells with pBabe-Empty or pBabe-GATA3 and then treated them with ionizing radiation (IR) or VP16. We found that after IR or VP16 treatment, the level of γ-H2AX in Gata3-overexpressing cells was significantly lower than the level of γ-H2AX in control cells (Fig. 5a, Additional file 1: Fig. S6a, S6c). Although overexpression of Gata3 induced the expression of CtIP, it is noteworthy that IR or VP16 treatment did not clearly alter its expression in Gata3-overexpressing cells (Additional file 1: Fig. S6b, S6c). In addition, we also confirmed that overexpression of Gata3 did not change the expression of RNF-8 and RAD51 in response to IR or VP16 (Additional file 1: Fig. S6b, 6c). These results suggest that Gata3 protects Brca1-deficient tumor cells from IR-induced DNA damage. We generated Gata3-overexpressing p18−/−;Brca1MGKO mammary tumor cells and transplanted them into the MFPs of mice. We found that tumors generated by Gata3-overexpressing cells were smaller in size, as previously reported [32], and displayed significantly less γ-H2AX-positive cells than tumors generated by control cells (Fig. 5b, c, Additional file 1: Fig. S7). Notably, Gata3-overexpressing tumors expressed drastically less mesenchymal markers, Vim and Fra1, than control tumors (Fig. 5c). Taken together, these data indicate that reconstitution of Gata3 in Brca1-deficient cancer cells restores the efficiency of DNA damage repair and suppresses mesenchymal differentiation in inhibition of tumorigenesis.
Fig. 5
Reconstitution of GATA3 in BRCA1-deficient tumor cells reduces DNA damage. a p18−/−;Brca1MGKO mammary tumor cells infected with pLVX-Flag (Empty) or pLVX-Flag-Gata3 (Gata3) were treated with or without ionizing radiation (IR) of 5 Gy, and the expression of Gata3 and γ-H2AX were analyzed at different time points after IR treatment. b, c p18−/−;Brca1MGKO mammary tumor cells infected with infected with pBabe-puro-Gata3 (Gata3) and pBabe-puro-empty (Empty) were transplanted into left and right inguinal MFP of mice. Four weeks later, the regenerated mammary tumors were analyzed by IHC with antibodies against γH2AX (b) Gata3, Fra1, or Vim (c). The H-scores for γH2AX in b were calculated. The results represent the mean ± SD of three individual tumors per group. The asterisk (*) denotes a statistical significance from empty- and Gata3-overexpressed samples determined by the T-test. The blots and images represent data from at least three independent experiments or tumors
GATA3 promotes HR-mediated DSB repair and restores HR efficiency of Brca1-deficient cellsDNA DSBs are repaired by two major pathways: HR and NHEJ. To directly test which pathway GATA3 is involved in DNA damage repair, we carried out assays for HR- or NHEJ-mediated repair using the DR-GFP/I-Sce I or EJ5-GFP/I-Sce I system [40]. We transfected pBabe-Gata3 (Gata3) or pBabe-Empty (Empty) in HR and NHEJ reporter cell lines, which were then infected with a virus expressing I-SceI. We found that overexpression of GATA3 significantly stimulated HR by more than twofolds when compared with the HR of control cells (Fig. 6a). However, overexpression of GATA3 had no significant effect on NHEJ (Fig. 6b). In line with these results, we found that knockdown of GATA3 significantly reduced HR, but not NHEJ (Fig. 6c, d), consolidating that GATA3 promotes DNA damage repair by HR, but not NHEJ. We then focused on the role of GATA3 in BRCA1-mediated HR. We found that KD of BRCA1 significantly reduced the level of HR and the expression of GATA3 (Fig. 6e), confirming not only the role of BRCA1 in promoting HR, but also our previous finding that BRCA1 regulates GATA3 in mammary cells [32]. Notably, we observed that the HR efficiencies in si-BRCA1 + GATA3 and si-Control + GATA3 cells were comparable, both of which were significantly higher than the HR efficiencies in si-BRCA1 + Empty cells (Fig. 6e, left). The results indicate that reconstitution of GATA3 restores the HR efficiency of BRCA1-deficient cells. Next, we checked the role of BRCA1 in GATA3-mediated HR. We found that the HR efficiency in si-GATA3 + BRCA1 cells was significantly higher than that in si-GATA3 + Empty cells, but slightly lower than that in si-control cells (Fig. 6f, left), suggesting overexpression of BRCA1 restores HR efficiency of GATA3-deficient cells. Interestingly, we detected that the expression of GATA3 in si-GATA3 + BRCA1 cells was drastically enhanced relative to that in si-GATA3 + Empty cells (Fig. 6f, right), indicating that overexpression of BRCA1 induced GATA3 re-expression in GATA3 knockdown cells, which is likely responsible for the restoration of HR efficiency of GATA3-deficient cells. These results support that GATA3 functions downstream of BRCA1 to promote HR.
Fig. 6
GATA3 promotes HR-mediated DNA damage repair and restores the efficiency of HR in Brca1-deficient cells. a, b The HR and NHEJ reporter cell lines, DR-GFP U2OS (a) and EJ5-GFP U2OS (b), were transfected with pBabe-Empty (Empty) or pBabe-GATA3 (GATA3), and then infected with virus expressing I-SceI. Two days later, cells were collected and analyzed for GFP expression by FACS (left panels) or for expression of GATA3 by qRT-PCR (right panels). c, d DR-GFP U2OS (c) or EJ5-GFP U2OS (d) cells were transfected with si-GATA3-1, si-GATA3-2 or si-control (si-Ctrl), infected with I-SceI, and analyzed for GFP (left) and GATA3 (right). The results in a–d represent the mean ± SD of three independent experiments. The asterisk (*) denotes a statistical significance from Empty and GATA3 or si-Ctrl and Si-GATA3 samples determined by the T-test. The number sign (#) denotes a statistical insignificance from Empty and GATA3 or si-Ctrl and Si-GATA3 samples determined by the T-test. e DR-GFP U2OS cells were firstly transfected with si-Ctrl or si-BRCA1 for 8 h and then transfected with pBabe-Empty (Empty) or pBabe-GATA3 (GATA3) for an additional 8 h. HR assay was done as above described (left) and gene expression was determined by qRT-PCR (right). f DR-GFP U2OS cells were firstly transfected with si-Ctrl or si-GATA3 for 8 h and then transfected with pBabe-Empty (Empty) or pBabe-BRCA1 (BRCA1) for an additional 8 h. HR assay was done as above described (left) and gene expression was determined by qRT-PCR (right). The results in e represent the mean ± SD of three independent experiments. The asterisk (*) denotes a statistical significance from Si-Ctrl + Empty and Si-Ctrl + GATA3, Si-BRCA1 + Empty, or Si-BRCA1 + GATA3 samples determined by the T-test. The asterisk (**) denotes a statistical significance from Si-BRCA1 + Empty and Si-BRCA1 + GATA3 samples determined by the T-test. The number sign (#) denotes a statistical insignificance from Si-Ctrl + Empty and Si-Ctrl + GATA3 samples determined by the T-test. The results in f represent the mean ± SD of three independent experiments. The asterisk (*) denotes a statistical significance from Si-Ctrl and Si-GATA3 + Empty samples determined by the T-test. The asterisk (**) denotes a statistical significance from Si-GATA3 + Empty and Si-GATA3 + BRCA1 samples determined by the T-test. The number sign (#) denotes a statistical insignificance from Si-Ctrl and Si-GATA3 + BRCA1 samples determined by the T-test
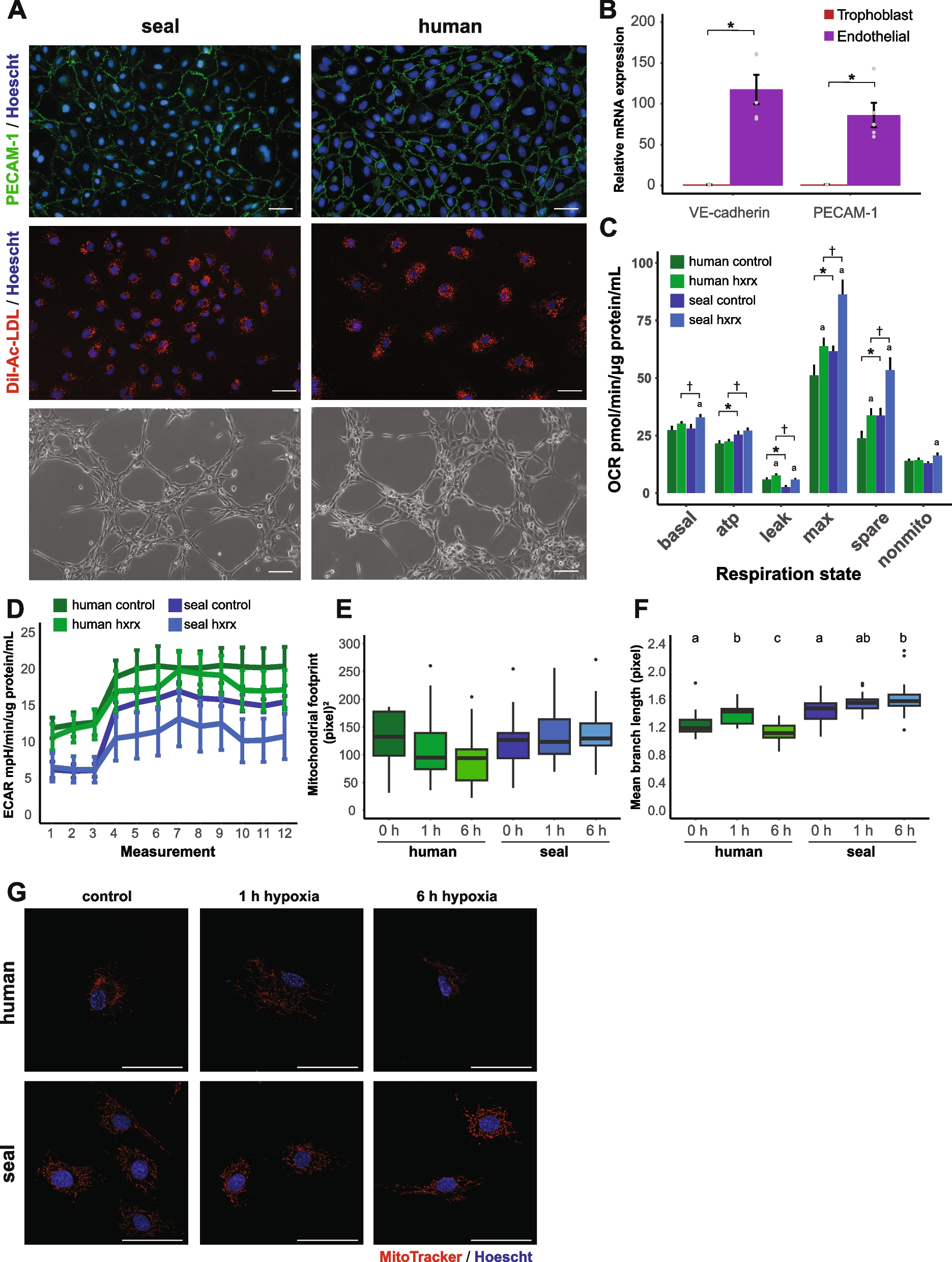
Depletion of Gata3 sensitizes tumor cells to PARP inhibitor, and reconstitution of Gata3 promotes resistance of Brca1-deficient tumor cells to PARP inhibitorWe have previously demonstrated that GATA3 is regulated by BRCA1 and GATA3 functions downstream of BRCA1 to control mammary cell differentiation [31, 32]. The findings that GATA3 stimulates DSB repair through HR and reconstitution of Gata3 restores the HR efficiency of Brca1-deficient cancer cells prompting us to investigate the response of Gata3-deficient tumor cells to PARP inhibitor. We treated Brca1+/− or Gata3+/− mammary tumor cells with olaparib (OLA), a PARP inhibitor. We noticed that the number of Brca1+/− or Gata3+/− tumor cells, but not the number of Gata3+/+;Brca1+/+ tumor cells, was significantly reduced after OLA treatment (Fig. 7a). In addition, OLA treatment of Gata3 knockdown tumor cells, not control cells, also resulted in a significant reduction of the cell number (Fig. 7b, c). These data support that depletion of Gata3, like that of Brca1, in mammary tumor cells impairs HR-mediated DNA DSB repair.
Fig. 7
Gata3-deficient tumor cells are sensitive to PARPi and reconstitution of Gata3 de-sensitizes Brca1-deficient cells to PARPi. a MMTV-PyMT (Gata3+/+;Brca1+/+), p18−/−;Gata3+/− (Gata3+/−), and p18−/−;Brca1+/− (Brca1+/−) mammary tumor cells were treated with DMSO or OLA at 10 μM. Forty-eight hours later, the cell number was counted. b, c Gata3+/+;Brca1+/+ mammary tumor cells infected with psi-LVRU6GP-control (sh-Ctrl) or psi-LVRU6GP-Gata3 (sh-Gata3), selected with puromycin, and analyzed for Gata3 expression (b). Gata3+/+;Brca1+/+-sh-Ctrl and Gata3+/+;Brca1+/+-sh-Gata3 cells in b were treated with DMSO or OLA for 48 h; the cell number was counted (c). d, e p18−/−;Brca1+/− (Brca1+/−) mammary tumor cells infected with pLVX-Flag (Empty) or pLVX-Flag-Gata3 (Gata3), selected with hygromycin, and analyzed for Gata3 expression (d). Empty- and Gata3-expressing Brca1+/−
留言 (0)