
記住我




In vitro studies were performed using the human keratinocyte cell line (HaCaT) and human dermal fibroblast cell line. Cells were purchased from the American Type Culture Collection (ATCC). They were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin (Gibco, Grand Island, NY, USA) at 37 °C in a 5% CO2 incubator. The cells were irradiated using a UVB cross-linker. Hs68 dermal fibroblasts were seeded in 35-mm tissue culture dishes at a density of 2 × 104 cells per tissue. Lipopolysaccharide (LPS) 10 µg/ml was used as a negative control group in the experiment to determine the anti-inflammatory effect and dexamethasone (DEX) 1 μm was used as a positive control group [25]. Retinoic acid 1% was used as a positive control for collagen (Col1A1), collagenase (MMP1, MMP9 and MMP13) the moisturizing (Has3) qRT-PCR [26].
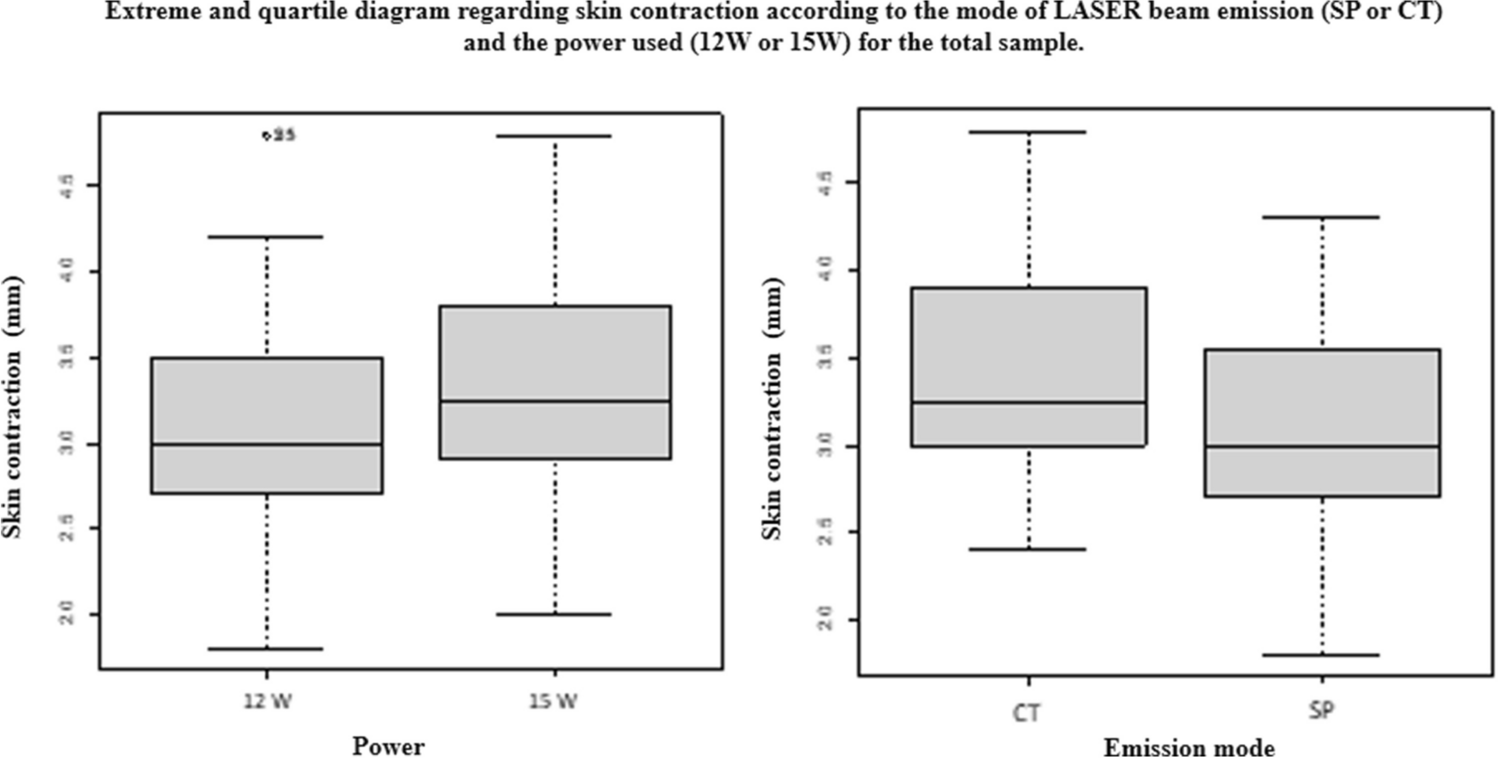
LED exposure in vitroThe Blue, White, NIR white, 680 nm chip, 720 nm chip, 4 chip (620 nm + 680 nm + 760 nm + 830 nm), 2 chip (660 nm + 850 nm) and nNIR irradiator used in this study was manufactured by Samsung Electronics Co., Ltd. The experimental LED illuminating systems, including LED with a white color and nNIR (780 nm centered phosphor), were installed in the cell culture incubator, which was maintained at a temperature of 37 °C and humidified atmosphere of 5% CO2. The spectrum for each wavelength is shown in Fig. 1 and Table 1. To reduce light interference, light was irradiated on the surface of the culture plate with partitions, which was 8 cm above the light source. The control group consisted of cells maintained in the dark. The irradiance of the LED sources was 6 mW/cm2, as measured using an MK350NPLUS LED meter (UPRtek, Taiwan). To check the effect of doses, we proceeded to each was performed according to the times and intensities indicated in Table 1. Then, cells were washed once with Dulbecco’s phosphate-buffered saline (DPBS, Welgene, Daegu, Republic of Korea) and exposed to 30 mJ/cm2 of UVB using a UV crosslinker (PCL-1000, BoTeck, Gunpo, Republic of Korea) without the culture plate cover.
Fig. 1
Spectrum profiles of the various LEDs
Table 1 Photobiomodulation parameter or LED parameter in vitroLED exposure of in vivoLight was irradiated at the top of the cage, which was 20 cm above the light source. The light exposure schedule was 30 min per day, three times a week for 2 weeks. The irradiances of the two light sources were the same at 6 mW/cm2 (Table 2).
Table 2 Photobiomodulation parameter or LED parameter in vivoCell cytotoxicity assayHs68 dermal fibroblasts and HaCaT keratinocytes were seeded in a 96-well plate at a density of 1 × 104 cells per well and cultured for 24 h. After washing with phosphate buffered saline (PBS), the cells were cultured for 24 h in medium containing fetal bovine serum (FBS). After removing the medium, cells were cultured in fresh medium without FBS for 24 h, and White and nNIR were irradiated under irradiation conditions of 10.80 J, 43.20 J, and 86.40 J. MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide; 0.5%) solution was added to the cultured 96-well plate. After incubation for 4 h, the culture medium was removed and 100 µL of dimethyl sulfoxide (DMSO) solution was added to dissolve MTT formazan. Then, absorbance was measured at 570 nm using a microplate spectrophotometer (BioTek, Winooski, VT, USA).
ROS measurement in Hs68 human dermal fibroblastsHs68 cells were seeded in a 96-well plate at a density of 1 × 104 cells per well and cultured for 24 h. After washing with PBS, cells were cultured for 24 h in medium containing FBS. The next day, the cells were treated with White and nNIR, which were 10.80 J, 43.20 J, and 86.40 J. After washing with PBS, 100 µL of DCF-DA solution diluted in the medium without phenol red and FBS was added and incubated at 37 °C for 30 min. After washing again with PBS, 100 µL of H2O2 (900 µM) solution diluted in the medium without phenol red and FBS was added and reacted in an incubator at 37 °C for 30 min. After washing with PBS, the cells were transferred to a 96-well black-bottom plate, and fluorescence intensity was measured using a microplate spectrophotometer.
ATP assay in vitro and in vivoBased on the principle of color development, the amount of ATP produced in Hs68 dermal fibroblasts, HaCaT keratinocytes, or mouse serum was measured using an ATP assay by following the instructions of the assay kit (Abcam, Cambridge, UK). After adding ATP assay buffer to the cell pellet or mouse serum and centrifuging at 4 °C and 13,000 rpm for 5 min, the supernatant was collected in a new tube. After adding cold 1 N perchloric acid solution, interfering proteins were removed for 15 min at 4 °C. After centrifugation at 13,000 rpm for 5 min, the supernatant was collected into a new tube. After adding cold 1 N sodium hydroxide solution, the mixture was allowed to react at 4 °C for 5 min. ATP reaction buffer was added after mixing the 96-well plate with the above solution well with standards and samples; light was blocked, and the plate was incubated at room temperature for 30 min. Thereafter, the fluorescence intensity was measured using a microplate spectrophotometer (excitation, 485 nm; emission, 535 nm) of the 96-well plate on which the reaction was completed.
qRT-PCRTotal RNA was extracted from either cell line or dorsal skin using Trizol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. The single-stranded cDNAs were synthesized using the Prime Script 1st strand cDNA Synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA). All cDNA templates were mixed with SYBR Green Master Mix (Bioneer, Seoul, Korea), and qRT-PCR was performed in an ABI 7700HT thermal cycler (Thermo Fisher Scientific, Waltham, MA, USA). The mRNA cycle threshold (Ct) values were normalized against 36B4 for ΔCt in the same sample, and then to the control sample to produce ΔΔCt. Finally, the fold-change (2 −ΔΔCt) was calculated. Primer sequences for each gene are listed in supply Table 3.
Table 3 Specific primer sequences of genePhotoaging in vivo mouse modelAll animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the Seoul National University Bundang Hospital (IACUC No. BA-2205-343-003-02). The experimental animals used in this study were 7-week-old SKH-1 hairless mice purchased from Orient Bio Co., Ltd., and bred at 20–24 °C, 40–60% humidity and 60 dB or less noise in a 12-hour light-dark cycle. The experimental animals were used after a 1-week acclimatization period, and anesthesia was induced using respiratory anesthesia while maintaining the concentration of isoflurane at 3%. When anesthesia was confirmed, UVB irradiation was used to induce photoaging. In the positive control group, 200 µL of retinoic acid (0.83 mM) was applied to the skin of the back area of mice thrice a week for 2 weeks. In the test group, White and NIR LED were irradiated to the skin of the back area thrice a week for 2 weeks, and the total irradiation amount was 64.80 J/cm2. Thereafter, visual evaluation of the experimental animals was carried out at the predetermined time points (day 0 and day 14 after UVB irradiation), and euthanasia was performed in a CO2 gas chamber 42 days after the start of the experiment. The skin on the back of the experimental animals was biopsied, and the skin tissue was fixed in 10% formalin solution to produce paraffin blocks.
Western blot analysis and measurement of type I procollagenTotal proteins were extracted from the dorsal skin in RIPA lysis buffer supplemented with complete protease inhibitor cocktail (GenDEPOT, Barker, USA) following the standard protocol (2). The supernatants were collected after centrifugation at 13,000 rpm at 4℃ for 30 min. Protein concentration was determined using a Bradford assay (Bio-rad, Hercules, CA). For western blot analysis, Equal amount of denatured proteins were separated by SDS-PAGE method and transferred to the nitrocellulose membrane. After blocking non-specific binding by incubation with 5% non-fat milk for 1 h, the membrane was incubated with a primary MMP13 antibody (company) overnight at 4℃ and followed by incubation with HRP-conjugated secondary antibody at 37 ℃ for 2 h. The band was visualized using ECL system (DonginBiotech, Seoul, Korea). Using the ImageJ software, we quantitatively evaluated the gray integration values of each band. Type I procollagen was measured according to the manufacturer’s protocol by the type I procollagen kit (Takara, Tokyo, Japan) and evaluated at 450 nm using microplate reader (BioTek, Winooski, VT, USA).
Hair growth assay in vivo mouse modelAll animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the Seoul National University Bundang Hospital (IACUC No. BA-2205-343-002-01). The experimental animals used in this study were 7-week-old C57BL/6NCrlOri mice (Orient Bio Co., Ltd., KR) that were bred at 20–24 °C, 40–60% humidity60 dB or less noise, in a 12-h light-dark cycle. The experimental animals were used after a 1-week acclimatization period, and anesthesia was induced using respiratory anesthesia while maintaining the concentration of isoflurane at 3%. White and nNIR were irradiated three times a week for 3 weeks, and the total irradiation amount was 97.2 J. Visual evaluation was performed at the time points (days 0, 7, 14, and 21 after hair removal), and euthanasia was performed in a CO2 gas chamber on day 21 after hair removal. Visual evaluation (photography) and histological evaluation for hair improvement were performed by applying 3% minoxidil or a test light source to mice (C57BL/6NCrlOri, 7-week-old).
Skin thickness analysisIn this study, the skin tissues of euthanized experimental animals were fixed with a 10% formalin solution, paraffin blocks were prepared, and tissue slides were prepared by sectioning them to a thickness of 3 μm. After hydration, a hematoxylin and eosin (H&E) solution was used for staining [27]. Skin thickness was measured as the distance from the top of the epidermis to the bottom of the dermis using Image J software.
Anagen induction scoreAn anagen induction assay was performed as previously described [28]. On performing histological analysis via H&E staining, anagen induction scores were calculated using an assigned arbitrary score (telogen = 1, anagen I-VI = 2–7), and the mean score was compared between the control mouse groups.
Statistical analysisStatistical analyses were performed using SPSS software (IBM, Armonk, NY, USA). Normality was verified using the Wilcoxon signed-rank test. Statistical analysis of the variables for parametric values was performed using a paired t-test. Statistical significance was set at p < 0.05 indicated using *, p < 0.01 indicated using **, and p < 0.001 indicated using *** (p < 0.05, **p < 0.01, ***p < 0.001).
留言 (0)