Human samples
Villus and decidual samples were obtained from patients with uRPL (n = 39) and those undergoing elective IA (n = 85) at the Department of Obstetrics and Gynecology of Tangdu Hospital. Donor characteristics are shown in Tables S1 and S2. The diagnosis of abortion was according to the guidelines of Tangdu Hospital, such as no fetal heartbeat for 1 week and reduced serum β-human chorionic gonadotropin and progesterone levels. Fetal cardiac activity was assessed using Doppler ultrasound at 6–8 weeks of gestation. The inclusion criteria for the uRPL group were as follows: 1) history of two or more consecutive spontaneous abortions, excluding ectopic pregnancies and molar pregnancies, including biochemical pregnancy; 2) age under 35 years and gestational age between 6–8 weeks.; and 3) normal villus chromosome karyotype. The exclusion criteria were as follows: 1) uterine structural malformations such as uterine septum, uterine myoma, and adenomyosis; 2) endocrine diseases such as diabetes, hyperprolactinemia, and hypothyroidism; 3) reproductive tract infections including chronic endometritis; 4) parental chromosomal abnormalities; and 5) a history of autoimmune diseases and thrombophilia. For the control group, age-matched patients or women with no previous pregnancy loss who underwent voluntary IA at 6–8 weeks of gestation were recruited. None of the women had a history of spontaneous abortion, pre-eclampsia, or preterm birth. The exclusion criteria were the same for patients with uRPL. All specimens were obtained after obtaining written informed consent from patients. This study protocol was approved by the Ethics Committee of Tangdu Hospital (TDLL2018-03–39).
Preparation of exosome-depleted FBS
Exosome-depleted serum was prepared as previously described [25]. Briefly, FBS was centrifuged at 180,000 × g for 18 h (Optima XE-100 Ultracentrifuge, SW32Ti rotor, Beckman Coulter) and 80% of the upper layer of FBS was collected and then filtered through a 0.22 μm filter (Millipore).
Villus explant culture
Fresh villus samples were obtained from the operating room and transported to the laboratory on ice within 10 min. Villus samples were washed in cold 1 × PBS (Corning), minced into 1 mm pieces, and transferred to Netwell™ inserts (Corning) in DMEM/F12 medium supplemented with 2% exosome-depleted FBS, 5 ng/mL epidermal growth factor (MCE), 1 × Insulin-Transferrin-Selenium Solution (MCE), 400 U/L hCG (Lizhu), 100 μg/mL streptomycin and 100 U/L penicillin (Millipore). Explants were incubated under 5% CO2 at 37℃ for 16 h. The same batch of FBS was used in all experiments. After incubation, the villus explants were centrifuged at 5,000 × g for 30 min to densify the pellets, which were then weighed after removing all traces of culture medium.
Isolation and characterization of villus-derived exosomes
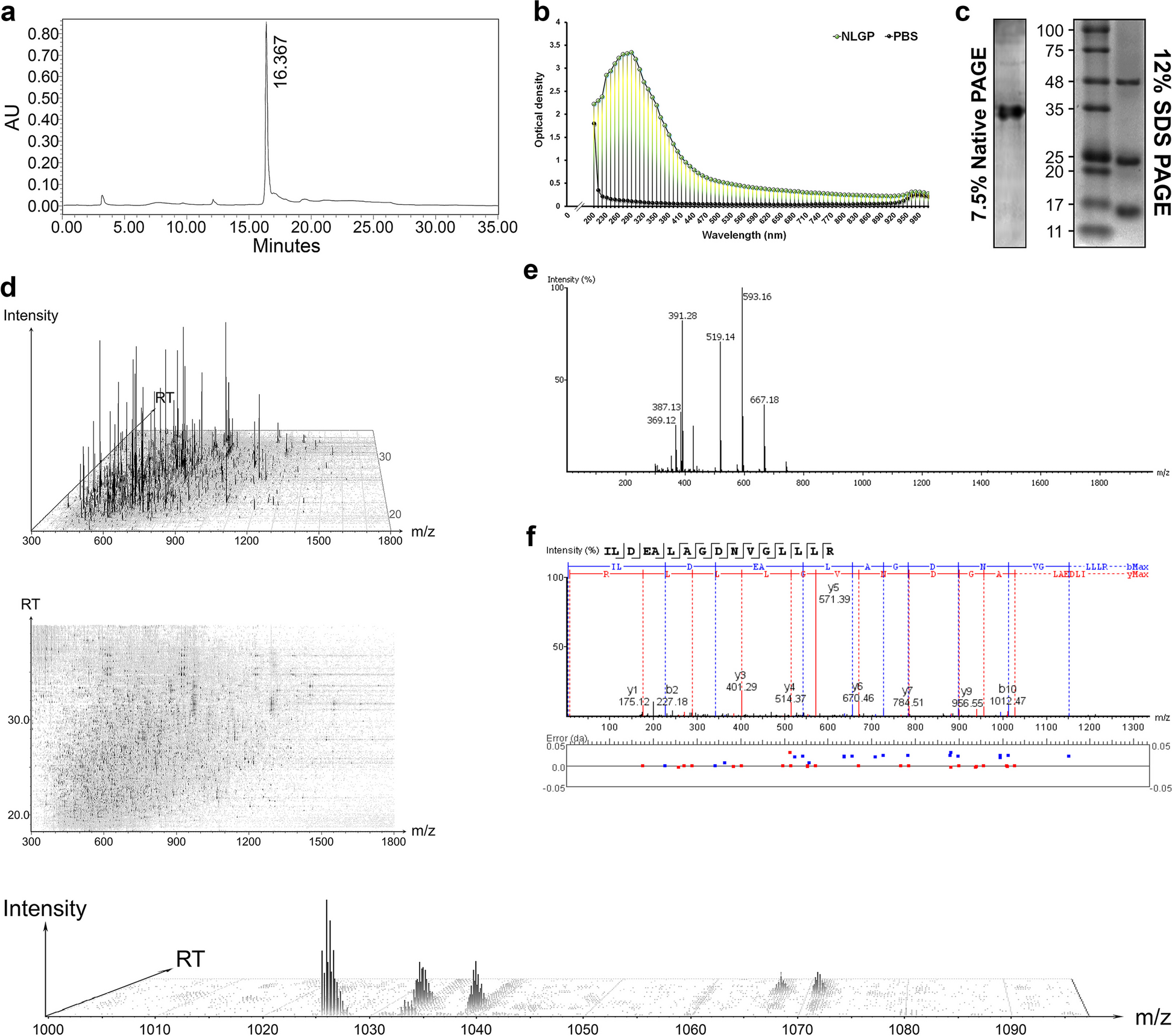
Exosomes were isolated from villus explant culture supernatants by sequential centrifugation. First, the supernatant was centrifuged (Hermle, Z446K) at 500 × g for 10 min to remove tissue explants, followed by centrifugation at 3,000 × g for 15 min and 20,000 × g for 2 h to remove dead cells and cell debris. The remaining supernatant was ultracentrifuged (Eppendorf, CP80NX) at 120,000 × g for 90 min and the pellet was washed with cold PBS and ultracentrifuged again. The final exosome pellets were resuspended in 100 μL of PBS and stored in low protein-binding Eppendorf tubes at 2–8℃ for 1 week and at -80℃ for long-term storage. All of the steps above were performed at 4℃ in a laboratory environment.
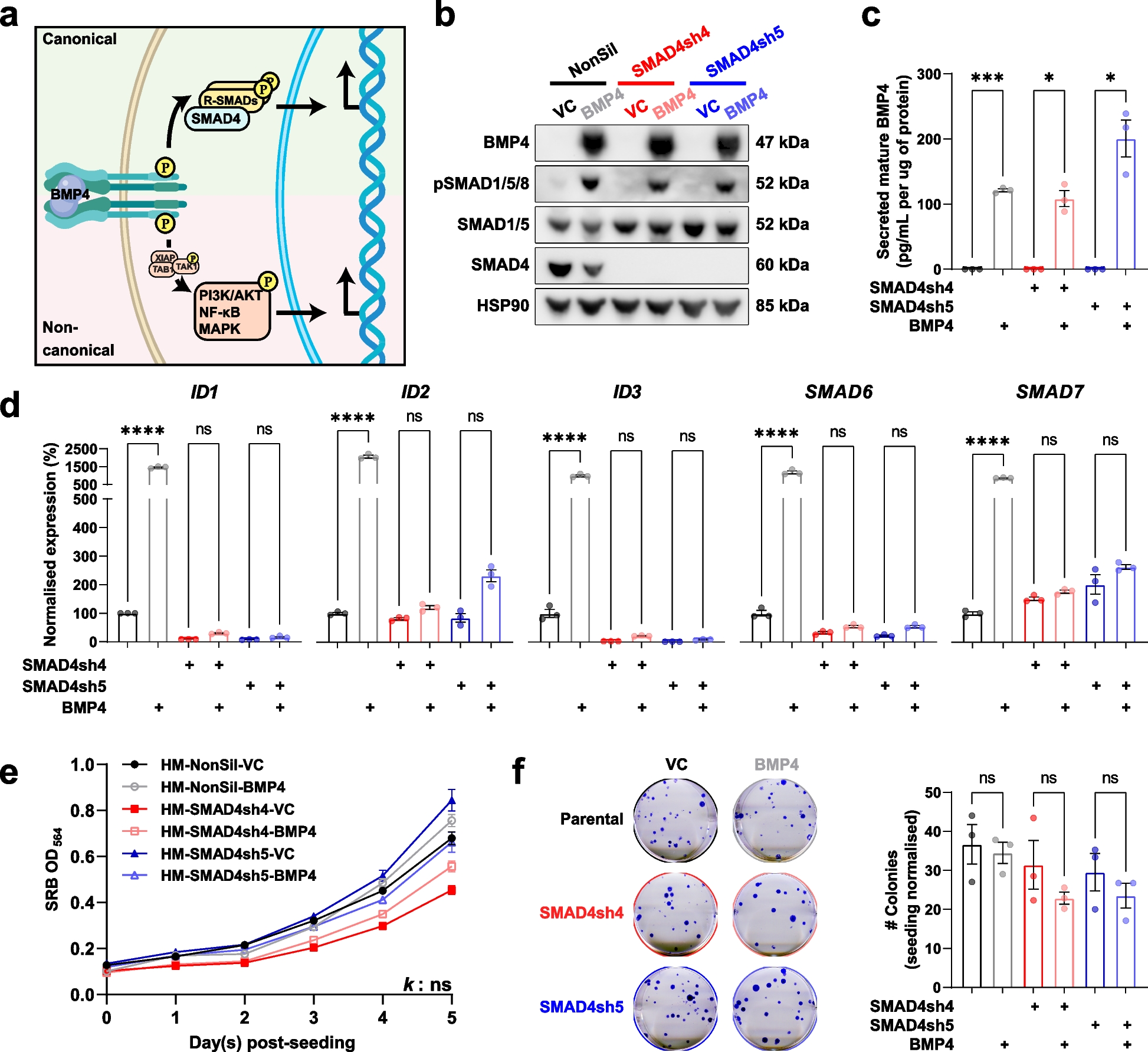
Nanoflow cytometry was used for size distribution analysis and exosome morphology was determined using transmission electron microscopy (TEM). These two procedures were performed by LC-Bio-Technologies (Hangzhou) Co., Ltd., as previously reported [26]. Exosomal markers were identified by immunoblotting, as previously described [27]. Exosome surface markers were detected by immunoblotting using anti-CD9 (ab263019, 1:1,000; Abcam), anti-CD63 (ab134045, 1:2,000; Abcam), anti-calnexin (ab133615, 1:2,000; Abcam), and anti-HLA-G (ab52455, 1:2,000; Abcam) antibodies. To measure the protein levels of exosomes, a bicinchoninic acid (BCA) kit (Thermo Scientific) was used according to the manufacturer’s instructions.
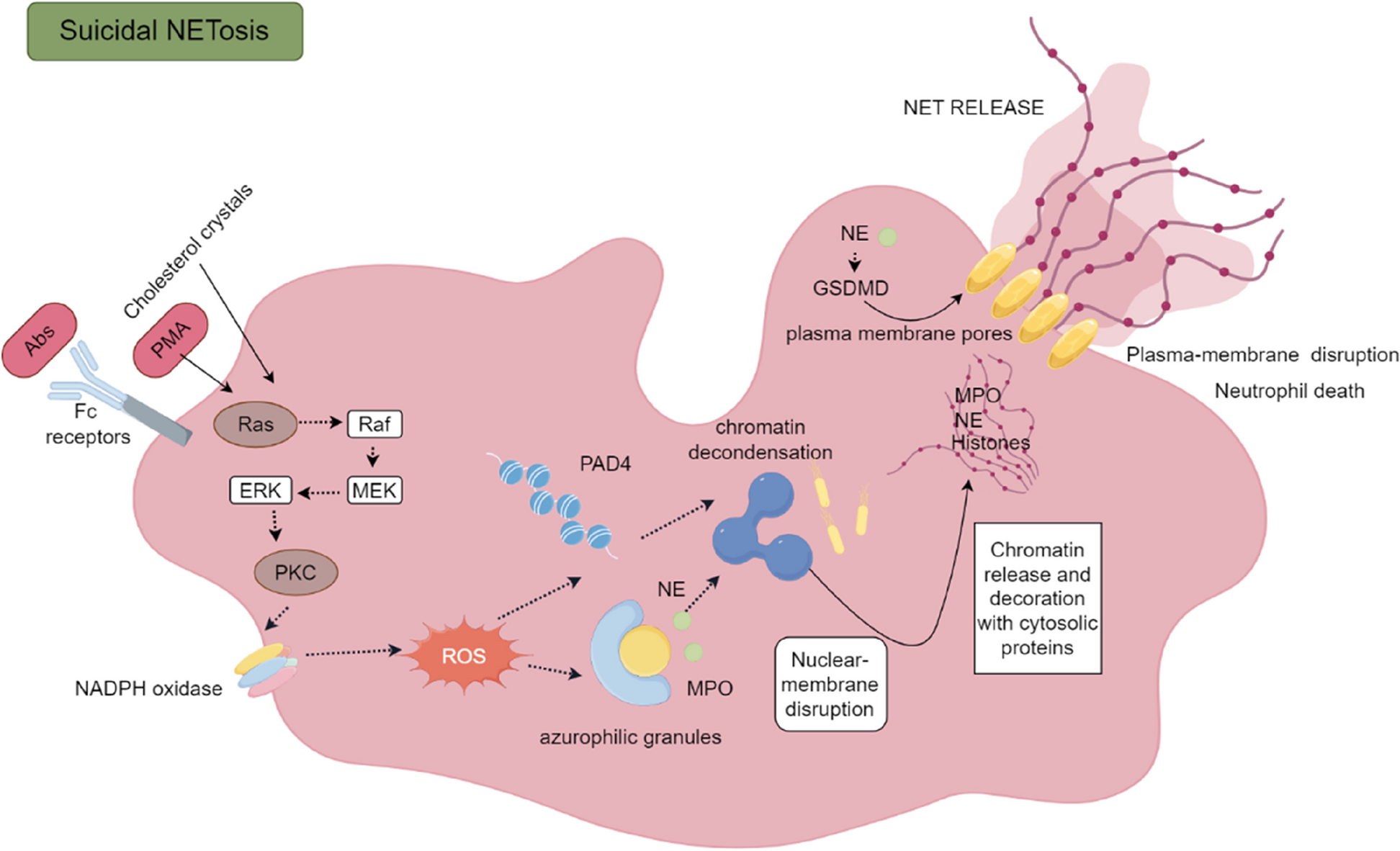
Isolation of human dNK cells
Fresh decidual samples were obtained from the operating room and transported to the laboratory on ice within 10 min. Decidual samples were washed with cold 1 × PBS (Corning) and minced into 1 mm pieces. The tissue pieces were then transferred to a 50 mL centrifuge tube (Corning) and digested with 1 mg/mL collagenase type IV (Sigma-Aldrich) and 0.025 mg/mL DNase I (Sigma-Aldrich) in RPMI 1640 medium (Gibco) for 1 h in a 37℃ water bath with continuous shaking. The suspensions were strained through 100 μm, 70 μm, and 40 μm nylon mesh (Falcon) and centrifuged at 220 × g for 10 min. Erythrocyte lysis buffer (Miltenyi Biotec) was used to remove erythrocytes from the collected cells. The collected cells were washed twice with RPMI-1640 and resuspended in 50 mL RPMI-1640 medium containing 10% FBS (Corning), 1 × non-essential amino acids, 1 × GlutaMAX and 1% penicillin–streptomycin (all from Gibco). The cells were cultured in T75 cell culture flasks for 2 h at 37℃ in a humidified 5% CO2 incubator (Thermo) to remove decidual stromal cells and macrophages. Only non-adherent cells, also called decidual immune cells, were collected and washed twice with complete medium. NK cells were further purified by negative selection using a magnetic-activated cell sorter (Miltenyi Biotec). The purity of the resulting dNK cells was > 94% CD56+. To simulate the in vivo state of dNK cells in patients, 0.05 ng/mL recombinant human IL-12 protein (R&D Systems, 10,018-IL) was added to the medium.
Exosome labelling with PKH26
Exosomes (500 μg) were incubated with 100 μL PKH26 working solution (Sigma) at 25℃ for 10 min away from light. After incubation, 5 mL of 1 × PBS was added to terminate labeling, and the exosome isolation protocol was repeated to remove free dyes.
Co-culture of exosomes and dNK cells
For incubation with primary dNK cells, purified exosomes were resuspended in complete RPMI-1640 medium to a final concentration of 100 μg/mL. The same volume of PBS was added to the medium as a negative control. The mixtures were exposed to purified dNK cells and incubated for 12 h or longer.
Enzyme-linked immunosorbent assay (ELISA)
Concentrations of IFN-γ in dNK cell supernatants or tissue protein extracts were quantified using a human IFN-γ ELISA kit (R&D Systems), which was used according to the manufacturer’s instructions.
RNA isolation and RT-qPCR
Long RNAs (> 200 bp) were extracted from purified human decidual NK cells treated with different interventions, using an RNA purification kit (Thermo Scientific). We extracted miRNAs from decidual immune cells and exosomes using RNAiso as a small RNA reagent (TaKaRa). Reverse transcription of long RNAs was performed using PrimeScript™ RT Master Mix (TaKaRa) and Mir-X™ miRNA FirstStrand Synthesis reagent (TaKaRa) was used for miRNA. miRNA expression levels were normalized to the external reference gene cel-miR-39 (Qiagen), a nematode miRNA that is not expressed in humans or mice [28]. The resulting cDNA was analyzed for expressed genes by real-time quantitative RT-qPCR with the One Step TB Green PrimeScript RT-PCR Kit II (TaKaRa Bio) or TB Green Advantage qPCR Premix (TaKaRa Bio). PCR primers used in experiments here are listed in Table S5.
Flow cytometry
Flow cytometry was performed according to a protocol provided by Biolegend.
Tube formation assay
Tube formation assays were performed as described previously [29]. Briefly, conditioned media were collected from treated dNK cells supernatants and centrifuged at 20,000 × g, 37℃ for 2 h. Then, 1.2 × 105 of human umbilical vein endothelial cells (HUVECs) were resuspended in different dNK cell-conditioned media, seeded into 24-well plates pre-coated with 300 μL Matrigel (R&D Systems), and incubated at 37℃ for 6 h. Finally, the cells were photographed using a 10 × brightfield objective lens (Olympus IX73). At least three different fields of view were captured per well, and the experiment was independently repeated three times.
Exosomes electroporated with miRNA
Exosome electroporation was performed according to a protocol provided by Bio-Rad. A mixture of 0.5 OD miRNA (cel-miR-39, miR-29a-agomir/antagomir, GenePharma) and 200 μg exosomes were transferred to a 0.2 cm-gap electroporation cuvette and placed on ice. The electroporation system was set at 350 V, 150 mF, with two pulses. After electroporation, the samples were placed on ice for 30 min, incubated with RNase to remove non-loaded miRNAs attached to the exosome surface, and used for subsequent experiments.
Development and characterization of exosomes mixed with HA-Gel
Modified exosomes were added to commoditized HA-Gel (5 mg/mL; Bioregen, Co., Ltd., China, approved by the CFDA as a medical device [No. 20153641542]) at a ratio of 3:7 (w/w). To evaluate viscosity and injectability, we extruded the gel slowly through a 29G syringe needle, and a DHR-2 (TA Instruments) rheometer was used to evaluate rheological behavior. Furthermore, vEXOs-HA-Gel was incubated at 37 °C under a 5% CO2 atmosphere and weighed for seven days. Saturated rhodamine B (Mackun) solution was mixed with HA-Gel to the same concentration as modified exosomes and then the mixture was incubated in PBS (Hyclone) at 37 °C in a 5% CO2 atmosphere. The supernatant was removed and fresh PBS was added daily. The concentration of rhodamine B released into the PBS was then determined by measuring the optical absorbance at 554 nm using a full-wavelength microplate reader (Molecular Science).
Exosome labelling with DiR and in vivo tracking
Exosomes (approximately 2 μg/μL at the protein level) were incubated with 100 μM DiR (Umibio) at a ratio of 10:1 (v/v) at 37℃ for 30 min. After incubation, 10 mL of 1 × PBS was added to terminate labelling, and the exosome isolation protocol was repeated to remove free dyes.
Considering that mice gradually gain weight during pregnancy, each mouse was tail-vein injected with approximately 5 μg of DiR-labeled exosomes per gram of body weight according to the indicated treatment plan. The distribution of DiR-labeled exosomes in the whole body and major organs of the mice was detected using an IVIS Lumina II in vivo imaging system (Xenogen) after injection or uterine horn injection.
dNK cells transfected with miRNA
Lipofectamine™ RNAiMAX transfection reagent (Invitrogen) was used to transfect miRNA into dNK cells according to the reverse transfection protocol provided by Invitrogen. Briefly, an miRNA oligo or transfection reagent was diluted in Opti-MEM I Reduced Serum Medium (Gibco) and incubated at room temperature for 5 min to prepare RNAi duplex-Lipofectamine RNAiMAX complexes. The complexes were added to each well with an appropriate number of dNK cells and incubated for 24 h at 37℃ in a CO2 incubator. After incubation, the dNK cells were collected for RNA extraction and flow cytometry. The RNA oligo sequences used in these experiments are listed in Table S5.
Luciferase reporter-gene assay
The full-length wild-type or mutant 3′ UTR of IFN-γ was cloned into firefly luciferase reporter plasmids (Promega). HEK293T cells were co-transfected with a mixture of firefly luciferase reporter plasmids, pRL-TK Renilla luciferase plasmid (Promega), and appropriate constructs or miRNAs (GenePharma). Luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega) as previously described [30]. Transfection efficiency data were normalized to the ratio of firefly luciferase to Renilla luciferase activity. TargetScan was used to predict the binding site of miRNAs and 3′ UTR of IFN-γ according to a previous description [31].
In vivosafety evaluation
To investigate the safety of agomir-IA-vEXOs, major organ tissues were collected for histochemical analysis 24 h after the last administration. Organs were fixed in 4% paraformaldehyde for 48 h and embedded in paraffin. Each section was sectioned into 5 mm slices, processed for routine hematoxylin and eosin (H&E) staining, and visualized under a microscope (Nikon Ti).
Animals studies
Wild-type mice (BALB/c, DBA/2 J males, and CBA/J females) were purchased from Beijing Huafukang Biotechnology Co., Ltd., China. All mice were housed in a standard specific pathogen-free laboratory with free access to food and water, controlled temperature of 24℃ to 26℃, a humidity of 65%-70% and a simulated natural lighting cycle of 12 h from 07:00 to 19:00. All experimental procedures involving animals were conducted in accordance with the National Guidelines for Animal Usage in Research (China). Permission for animal studies was obtained from the Ethics Committee of Tangdu Hospital.
To construct the RPL mouse model and healthy control model, virgin female CBA/J mice were randomly introduced into DBA/2 J or Balb/c male mice, and the timing of conception was determined by detection of a vaginal plug as gestation days (GD) 0.5.
Uterine horn injection: The mice were operated on through the back approach, the uterine horn was gently pulled out, and the skin was sutured after slowly injecting liquid with a syringe. Mice were placed on a heat preservation board for 2 h and then returned to their cages. The mated mice were injected with 50 μL HA-Gel or PBS containing exosomes at GD 4.5 and GD 7.5. Sham and RPL groups were operated on and injected with PBS to exclude any influence of the operation.
Placental processing: Placental tissues were collected 24 h after the last administration, and each placenta was drained of surface liquid using absorbent paper and weighed. The placentas were randomly selected and used for subsequent analyses.
Statistics
Statistical analyses are summarized in the Figure legends. In most cases, the results represent means ± standard error of the mean (SEM) of n separate experiments. Two-tailed Student’s t-tests were used to compare two groups. Statistical significance was set at P < 0.05. One-way analysis of variance (ANOVA) was performed to determine whether there was a statistically significant difference between more than two datasets, followed by Bonferroni’s post-hoc correction.




留言 (0)