
記住我







Chlorophyll breakdown has been a very old enigma, and chlorophyll disappears without any noticeable traces (Hörtensteiner and Kräutler, 2011). This phenomenon is crucial catabolic process required for fruit ripening and leaf senescence. The recent identification of a chlorophyll breakdown mechanism that is highly conserved in land plants was made possible by the structural elucidation of colorless linear tetrapyrroles as final breakdown products of chlorophyll. The primary enzyme that opens the chlorin macrocycle of pheophorbide, giving all subsequent breakdown products their characteristic, is PAO. The necessity for a senescing cell to detoxify the potentially harmful pigment was used to explain the degradation of chlorophyll; however, new studies in leaves and fruits suggest that chlorophyll catabolites may have physiological functions (Jiao et al., 2020).
Degradation of chlorophyll causes a loss of green hue, which is the most obvious sign of leaf senescence. It has been believed that PAO is an essential enzyme in the breakdown of chlorophyll (Hörtensteiner, 2006). It ultimately creates the principal fluorescent chlorophyll catabolite (FCC) by oxygenolytically cleaving the porphyrin macrocycle of pheophorbide (pheide) (Xiao et al., 2015). PAO genes were first identified in maize (ZmLls1) (P. Li et al., 2006) followed by other species including rice (Tang et al., 2011), tomato (Spassieva and Hille, 2002), canola (Ho et al., 2006), wheat (Ma et al., 2012), and soybean (P. Li et al., 2006). Previous research demonstrated that environmental stressors and natural senescence in plants cause the production of PAO (Pruzinská et al., 2005). Lethal leaf spot 1 (LLS1) of maize and AtPAO, which is represented by the accelerated cell death 1 (ACD1) gene in Arabidopsis, are similar. It is a member of the tiny family of iron-sulfur oxygenases of the Rieske type (Pruzinská et al., 2003). Senescence caused in persistent darkness has been demonstrated to accumulate PAO and cause light-independent cell death in the absence of ACD1 (Tanaka et al., 2003; Hirashima et al., 2009). Recent researches on PAO have focused on the functional studies to inhibit the cell death. The Papper PAO gene express itself in response to different stressors and natural senescence. The CaPAO gene may be crucial in salt-induced leaf senescence and defense responses to a variety of stressors (Xiao et al., 2015).
The porphyrin macrocycle is opened by PaO, a nonheme iron monooxygenase found in the inner envelope of developing gerontoplasts, by introducing two oxygen atoms. Measurements of PaO activity have demonstrated that Pheide a is an effective substrate, while Pheide b functions as a competitive inhibitor (Ho et al., 2006). AtPaO is a five-member gene family in Arabidopsis that codes for nonheme iron oxygenases, which are distinguished by the coexistence of a mononuclear iron-binding domain and a Rieske-type domain. Additionally, this gene family contains Tic55, PTC52, choline monoxygenase, and chloroacetate oxygenase. This gene family also contains choline monooxygenase, Chl a oxygenase, Ptc52, and Tic55 (Gray et al., 2004).
Pears stand one of the most significant temperate fruit trees globally and belong to the Rosaceae family and the Amygdaloideae subfamily. Pears have been cultivated for over 3,000 years with 39 billion tons are delievered worldwide every year (Wu et al., 2014; 2018; Ferradini et al., 2017). Presently, 22 species of pear with 5000 accessions have been reported. Among these five species are majorly cultivated for fruit production, including Pyrus bretschneideri, P. pyrifolia, P. communis, P. ussuriensis and P. sinkiangensis (Li et al., 2022). The majority of cultivated pears have a diploid genome (2n = 34), which is extremely heterozygous and has several repeating sequences (S. Chen et al., 2023). In this project, eight pear genomes have been used; Cuiguan (P. pyrifolia), Shanxi Duli (P. betulifolia), Zhongai 1 [(P. ussuriensis × communis) × spp.], Nijisseiki (P. pyrifolia), Yunhong No.1 (P. pyrifolia), d’Anjou (P. communis), Bartlett v2.0 (P. communis), Dangshansuli’ v.1.1 (P. bretschneideri) have been used (S. Chen et al., 2023). Pear is an economical fruit with sweet taste and great nutritional value with a cultivation history of up to 3000 years back. However, no research is available on Pyrus PAO genes and their regulatory mechanism. Thus, this study provides information regarding the characterization of PAO gene family members from multiple Pyrus genomes, to understand their evolution, intra-specific, and functional diversity. Further, the expression profiles of Pyrus PAO genes in tissues, abiotic, and biotic stresses have been determined, providing valuable insights for future stress-resistant pear breeding. This comprehensive study will be useful for further functional investigations of Pyrus PAOs.
2 Materials and methods2.1 Identification of PAO genesThe protein sequences of Arabidopsis thaliana PAO proteins were obtained from The TAIR database (https://www.arabidopsis.org/). The protein sequence FASTA files of eight Pyrus species including Cuiguan, Shanxi Duli, Zhongai 1, Nijisseiki, Yunhong No.1, d’Anjou, Bartlett v2.0, Dangshansuli’ v.1.1 were used as subject sequences to run the command-line tool, BLAST+. A BLASTp search was conducted against the eight Pyrus proteomes using AtPAO protein sequences as the queries. The hits from this search were then refined, including the removal of isoforms and duplicates.
The Pyrus PAO candidate sequences were examined for domains through the NCBI Conserved Domain Database (CDD; https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) (Marchler-Bauer et al., 2011) and InterPro (https://www.ebi.ac.uk/interpro/) (Hunter et al., 2009) to identify the definitive protein family sequences. Physicochemical properties such as molecular weight (MW), isoelectric point (pI), aliphatic index (AI), instability index (II), and the grand average of hydropathicity (GRAVY) were forecasted using the ExPASy ProtParam tool (https://web.expasy.org/protparam/) (Gasteiger et al., 2005). The subcellular localization of these PAO proteins was forecasted using the WoLF PSORT tool (https://wolfpsort.hgc.jp/) (Horton et al., 2007).
2.2 Multiple sequence alignment, phylogenetic analysis, gene structure and conserved motifs analysisPhylogenetic analysis was carried out to assess the intra-specific evolutionary connections among Pyrus PAOs. A multiple sequence alignment of 11 Cuiguan, 8 Shanxi Duli, 10 Zhongai 1, 10 Nijisseiki, 10 Yunhong No.1, 9 d'Anjou, 5 Bartlett v2.0, 8 Dangshansuli’ v.1.1, 4 A. thaliana, 5 O. sativa, and 4 Z. mays protein sequences was performed using ClustalW (Thompson et al., 2003). The phylogenetic tree was constructed using the IQTREE Web Server (http://iqtree.cibiv.univie.ac.at/) (Trifinopoulos et al., 2016) with the maximum likelihood (ML) method and bootstrap replicates of 1000. The iTOL: Interactive Tree of Life (https://itol.embl.de/) (Letunic and Bork, 2007) was used for editing and visualization.
To identify the conserved common motifs in all eight Pyrus PAO sequences, the MEME tool (https://meme-suite.org/meme/) (Bailey et al., 2009) was utilized. The number of conserved motifs was set to 20 for each sequence. Gene structures were constructed using CDS and genomic sequences through the Gene Structure Display Server (GSDS; https://gsds.gao-lab.org/) (Hu et al., 2015). The identified motifs and gene structures were visualized using TBtools-II v2.067 (C. Chen et al., 2018).
2.3 Chromosomal mapping and duplication events analysisThe chromosomal positions of each Pyrus PAOs were extracted from GFF/GFF3 files and mapped to chromosomes using TBtools-II v2.067: Gene Location Visualize advanced (C. Chen et al., 2018). By assessing whether the shorter gene’s length covered 70% of the longer gene and if the alignment similarity between the two genes was 70% or higher, instances of PAOs gene duplication were identified (ul Qamar et al., 2023). The duplication pattern, whether segmental or tandem, was also examined. To predict the selection pressure on the duplicated genes, Ka/Ks values were calculated using DnaSP v.6 software (Le et al., 2011; Rozas et al., 2017). Based on whether the Ka/Ks ratio was greater than, equal to, or less than one, purifying, neutral, or positive selection was inferred (Zia et al., 2022). Additionally, the divergence time for the duplicated gene pairs was estimated using the formula “t = Ks/2λ×10–6″ in million years (Mya), where the λ value is 1.5 × 10−8 for dicots (Fatima et al., 2023; Sadaqat et al., 2023).
2.4 PPI and GO enrichment analysisThe amino acid sequences of the Pyrus PAOs proteins were analyzed for protein-protein interactions (PPIs) using the STRING database (Mering et al., 2003). The top ten interactions were selected for prediction, with an interaction threshold set at 0.4. The PPI network was then visualized using Cytoscape software (Shannon et al., 2003). Additionally, the PANNZER database (http://ekhidna2.biocenter.helsinki.fi/sanspanz/) (Törönen et al., 2018) was utilized to predict the GO enrichment analysis components, including biological processes (BPs), cellular components (CCs), and molecular functions (MFs).
2.5 Cis-regulatory elements prediction and expression analysis of pyrus PAOsTo predict the cis-regulatory elements, the 2 kb sequences upstream of the translation start site of Pyrus PAOs genes were obtained and analyzed using the PlantCARE database (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/) (Rombauts et al., 1999).
To understand the expression patterns of Pyrus Dangshansuli PAOs, transcriptomic RNA-seq data from different developmental stages of pear fruit (BioProject: PRJNA309745), under drought stress (BioProject: PRJNA655255), and Fruit hardening disease (BioProject: PRJNA763913) were retrieved from the SRA-NCBI database (https://www.ncbi.nlm.nih.gov/sra). The genome and annotation (GFF) files of Dangshansuli were downloaded from “The pear genomics database” (PGDB; http://pyrusgdb.sdau.edu.cn/) (S. Chen et al., 2023). The quality of reads was assessed using the FastQC tool (Wingett and Andrews, 2018). HISAT2 (Kim et al., 2019) was used to build the indexes of the Pyrus genome, and the high-quality paired-clean reads were then mapped onto the indexed genome. The abundance estimation of gene family members was carried out using StringTie (Pertea et al., 2016). Finally, a heatmap was generated using the Fragments Per Kilobase of transcript per Million mapped reads (FPKM) values (Zameer et al., 2021).
2.6 3D structure prediction of DaPAO proteinsTo ensure proper functionality, proteins require a three-dimensional (3D) structure. The 3D structures of eight Dangshansuli PAOs (DaPAO1-DaPAO8) were predicted using AlphaFold2 (https://alphafold.ebi.ac.uk/) (Jumper et al., 2021). The accuracy of these predicted structures was assessed through the SAVES server (https://saves.mbi.ucla.edu/) (Zameer et al., 2022) and MolProbity (http://molprobity.biochem.duke.edu/) (Davis et al., 2007). Finally, the UCSF ChimeraX software (Goddard et al., 2018) was utilized to visualize these structures (Neupane et al., 2018; Zia et al., 2024).
3 Results3.1 Identification of PAO genes in eight pyrus speciesA total of 11 genes from the Cuiguan genome (CuPAO), eight from Shanxi Duli (ShPAO), 10 from Zhongai1 (ZhPAO), 10 from Nijisseiki (NiPAO), 10 from Yunhong No.1 (YuPAO), nine from d’Anjou (AnPAO), five from Bartlett v2.0 (BrPAO), and eight from Dangshansuli’ v.1.1 genome (DaPAO) were identified. All of these members were confirmed to have the Rieske [2Fe-2S] iron-sulphur domain superfamily and Pheophorbide a oxygenase (PAO) domain. The protein names of each member were assigned based on their position on chromosomes (Table 1).
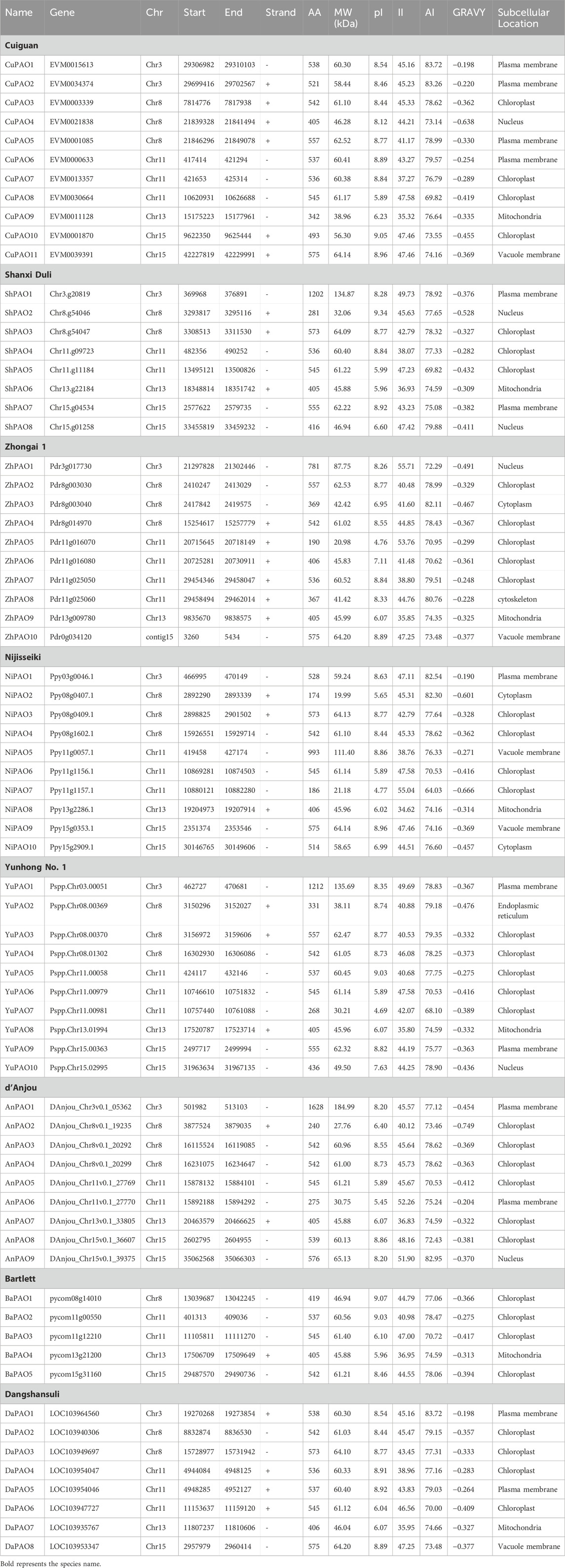
Table 1. The PAO gene family members identified in eight Pyrus species, their physicochemical characteristics, and subcellular localization.
The physical and chemical properties of all Pyrus PAO proteins were computed and compared. The protein length ranged from 174–1628 aa and molecular weight ranged from 19.99 to 18.50 kDa. The isoelectric point showed that most of the proteins are basic in nature. The instability index denoted that most of these proteins are unstable. Aliphatic index showed that proteins are good thermostable. GRAVY values indicates that the proteins have hydrophilic behaviour. Subcellular localization analysis revealed that most proteins were found in the chloroplast, with a few in the nucleus, plasma membrane, mitochondria (Table 1).
3.2 Phylogenetic relationships of pyrus PAO family membersTo examine the evolutionary connections among the PAO members from eight Pyrus genomes, a phylogenetic tree was created using 84 amino acid sequences from 11 species. All PAO proteins were grouped into four sub-family clusters: Group A, B, C, and D. The Group D cluster was the largest, with 24 members. After that the Group A was the second largest with 23 members. Group C and B has the members 22 and 15 respectively (Figure 1).
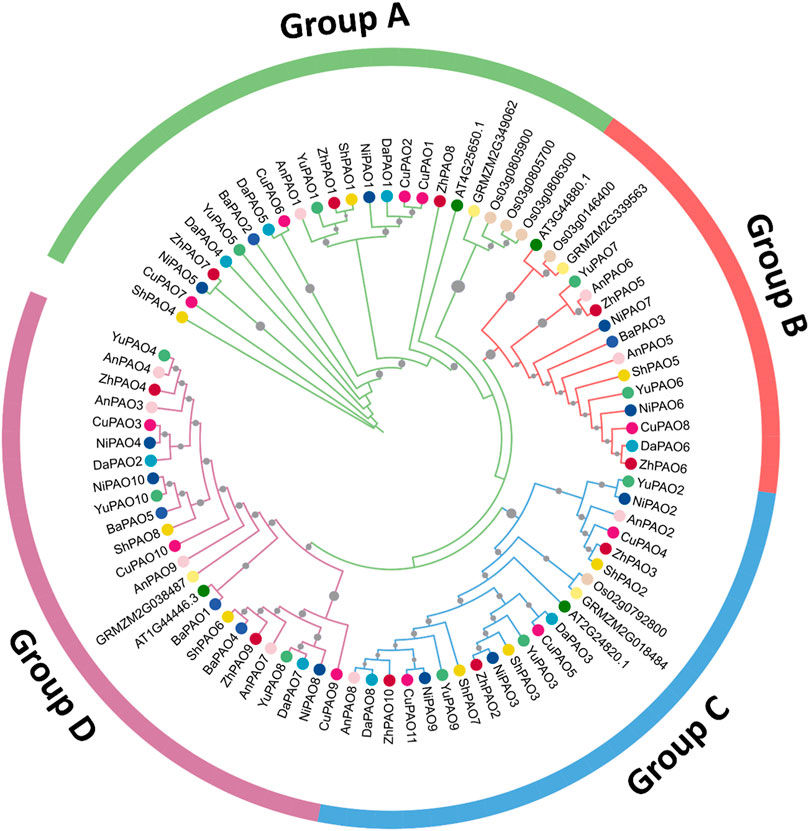
Figure 1. Phylogenetic tree of PAO gene family members from eight Pyrus, A. thaliana, O. sativa, and Z. mays genomes. Each group is represented by a particular color with specific symbol used for each species.
3.3 Gene structure and conserved motifs analysis of pyrus PAOsTo understand the evolutionary patterns among Pyrus PAOs, their conserved motifs and gene structures were studied. The gene structure was observed to be highly conserved within members of the same subfamily. The number of exons ranged from 3–18. Group A and B has more numbers of exon as compared to Group C and D (Figure 2A).

Figure 2. (A) Gene structure showing conservation pattern of exons and introns and (B) Pattern of conserved motifs.
All members of each subfamily shared highly conserved motifs. Members of Group A, B, C, and D had exactly the same motif pattern (conserved motifs 2, 3, 4, 5, 8, 11, 12, 14, 16). Motif seven was only present in Group D and the motifs 6, 9, 10, 13, 15, 17, and 18 were present in all members except few members of Group D (Figure 2B). This high conservation of motifs suggests no major differences in the structure and functions of Pyrus PAOs.
3.4 Chromosomal location and gene duplication analysisTo assess the gene distribution pattern of Pyrus PAOs across the 17 chromosomes of each Pyrus genome, their chromosomal gene localization was determined. This analysis revealed an uneven distribution of genes across chromosomes. In the Cuiguan genome, 11 CuPAOs were localized on five out of seventeen chromosomes (Chr3, 8, 11, 13, and 15). The remaining chromosomes did not contain any CuPAOs genes. Similarly, in the Shanxi Duli genome, eight ShPAOs were scattered across five out of seventeen chromosomes. In the Zhongai1 genome, 10 ZhPAOs were distributed across four out of seventeen chromosomes. In the Nijisseiki genome, 10 NiPAOs were distributed across four chromosomes. In the Yunhong No.1 genome, 10 YuPAOs were unevenly distributed across four chromosomes. In the d’Anjou genome, nine AnPAOs were distributed across five chromosomes. In the Bartlett v2.0 genome, five BaPAOs were distributed across four chromosomes. Finally, in the Dangshansuli’ v.1.1 genome, eight DaPAOs members were distributed across five out of seventeen chromosomes (Figure 3).
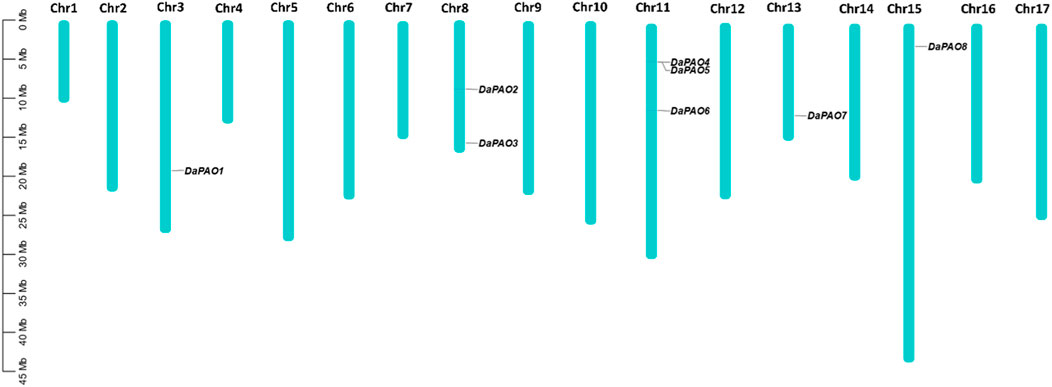
Figure 3. Chromosomal map showing eight DaPAOs distributed on five Dangshansuli’ v.1.1 chromosomes.
Gene duplication events were investigated within each Pyrus PAO gene family member. In the Cuiguan genome, eight pairs of genes were found to be duplicated, in which two pairs duplicated through tandem duplication and six pairs duplicated through segmental duplication. The Shanxi Duli genome contained two segmentally duplicated pairs of ShPAOs. In the Zhongai1 genome, three pairs of ZhPAOs were duplicated, with two pairs being tandemly duplicated and the one pair being segmentally duplicated. The Nijisseiki genome had two pairs of segmentally duplicated NiPAOs. In the Yunhong No.1 genome, three pairs of YuPAOs were found, all originating from segmental duplication. The d'Anjou genome exhibited four pairs of duplicated genes, with two pairs being tandemly duplicated and the other two pairs segmentally duplicated. The Bartlett genome contained only one pair of segmentally duplicated genes. The Dangshansuli genome had four pairs in which three pairs are segmentally duplicated and one gene pair is tandemly duplicated (Table 2).
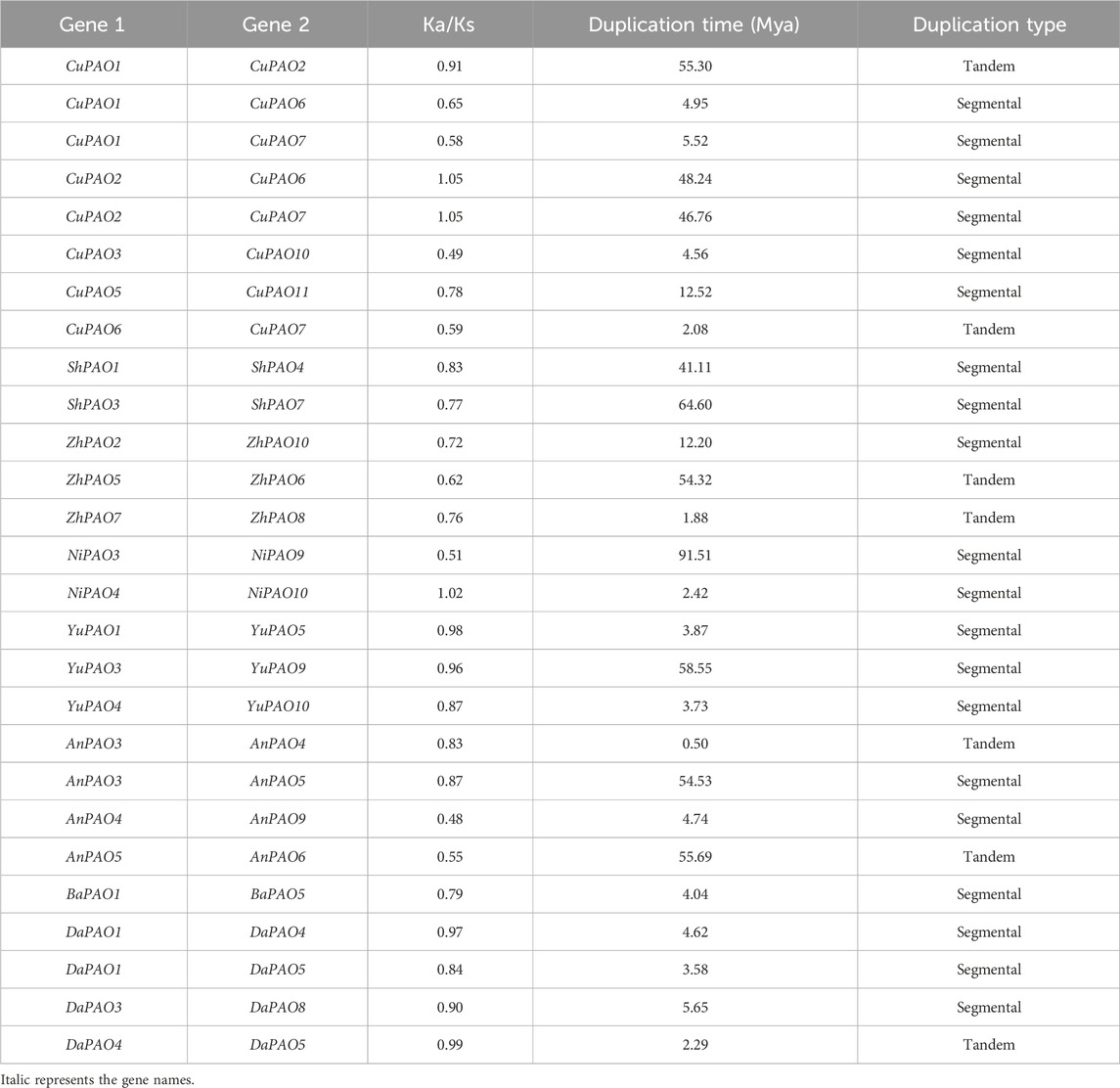
Table 2. Duplication data of Pyrus PAOs, rate of synonymous (Ka) and non-synonymous mutations (Ks), duplication time (Mya), and type of duplication.
To investigate the evolutionary pressures acting on the duplicated Pyrus PAOs genes, the Ka, Ks, and Ka/Ks ratios were computed for all para-homologous gene pairs. Across the eight genomes, the Ka/Ks ratio varied from 0.48 to 1.05, indicating a mix of positive and negative selection events. The divergence time of the 27 duplicated gene pairs of Pyrus PAOs ranged from 0.5 to 91.51 million years ago (MYA) (Table 2).
3.5 PPI and GO enrichment analysisA protein-protein interaction (PPI) network of the Pyrus PAOs was constructed to explore the functional diversity among its members. Among the five Dangshansuli members, DaPAO2, DaPAO5, DaPAO6, DaPAO7, and DaPAO8, interactions with several other proteins were observed. The majority of these interactions were identified with PORA (degree 9) and NYC1 (degree 9) proteins, suggesting the potential roles of these Pyrus members in skotomorphogenesis, photomorphogenesis and throughout the plant life under specific light conditions (Figure 4A).

Figure 4. (A) Interactions among Pyrus PAOs and other homologous proteins. (B) Predicted BPs, CCs, and MFs associated with Pyrus PAOs.
GO enrichment analysis was conducted to determine the molecular functions of Pyrus PAOs in a dynamic context. This analysis classified the genes into three main categories: biological processes (BPs), cellular components (CCs), and molecular functions (MFs). The primary BPs identified included the protein targeting to chloroplast, chlorophyll biosynthetic process, and flower and fruit Development. The genes were found to be present in CCs such as the chloroplast, plastid envelope, and thylakoid membrane. The MFs associated with these genes included 2 Iron, two sulfur cluster binding, metal ion binding, and pheophorbide A oxygenase activity (Figure 4B, Supplementary Material S1).
3.6 Cis-regulatory element analysis of pyrus PAOsTo gain a deeper understanding of the diverse stress responses exhibited by Pyrus PAOs, the cis-regulatory elements in their promoter sequences were analyzed. Across all genomes, cis-elements associated with stress responses, including light, hormones, and development, were found in abundance. Specifically, G-box, GT1-motif, and GATA-motif (cis-element Box 4) were identified as being involved in the regulation of light-stress. Hormone responsiveness was linked to five cis-elements: P-box, TGA-element, ABRE, CGTCA-motif, and TCA-element. Additionally, the GC-motif, LTR, TC-rich repeats, and MBS were identified as the four cis-elements associated with stress responsiveness. For developmental processes, five elements were involved: CAT-box, MBSI, circadian, HD-Zip 1, and o2-site. The presence of these elements in Pyrus PAO suggests their involvement in hormone, stress, and development-related responses (Figure 5, Supplementary Material S2).
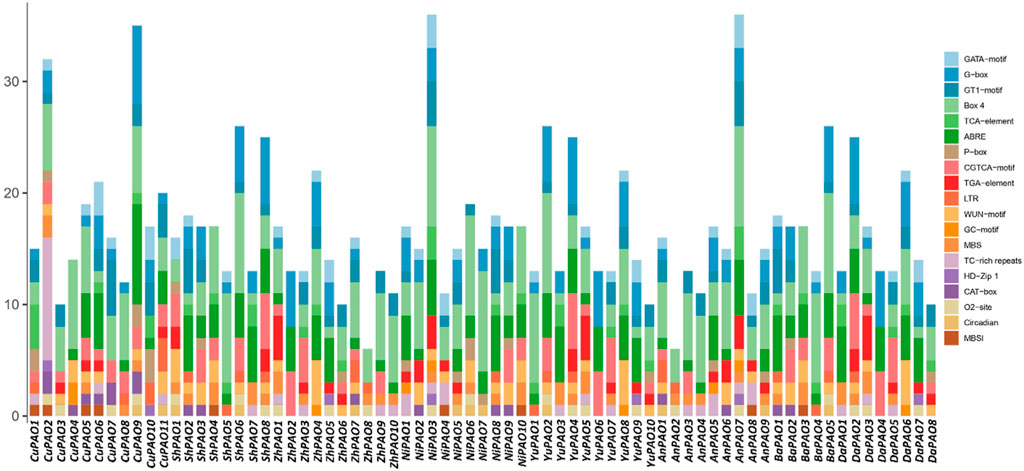
Figure 5. The Pyrus PAOs genes’ upstream promoter regions contain cis-regulatory elements. Each bar represents a distinct element found in a given gene.
3.7 Gene expression profiling of pyrus PAOsTranscriptome expression data were utilized to assess the expression levels of eight DaPAOs under fruit hardening disease to evaluate the expression of DaPAOs in diseased conditions. DaPAO4 and DaPAO5 were highly expressed in all conditions. DaPAO1 was downregulated in disease conditions as compared to normal condition (Figure 6A). Under drought stress, different expression levels were observed, with DaPAO2 being upregulated. On the other hand, the DaPAOs have no change in expression during normal and drought condition (Figure 6B). Transcriptome expression data was also utilized to assess the expression levels of eight DaPAOs across various tissues, including fruit, leaves, petal, sepal, ovary, stem, and bud. DaPAO1 was highly expressed in ovary. Three genes named as DaPAO2, 3, and 8 were highly expressed in stem. Two genes named as DaPAO5 and DaPAO6 were highly expressed in most of the tissues (Figure 6C). DaPAO1, DaPAO3, DaPAO4, and DaPAO5 showed fluctuated and high expression patterns which make these genes potential candidates for future research.
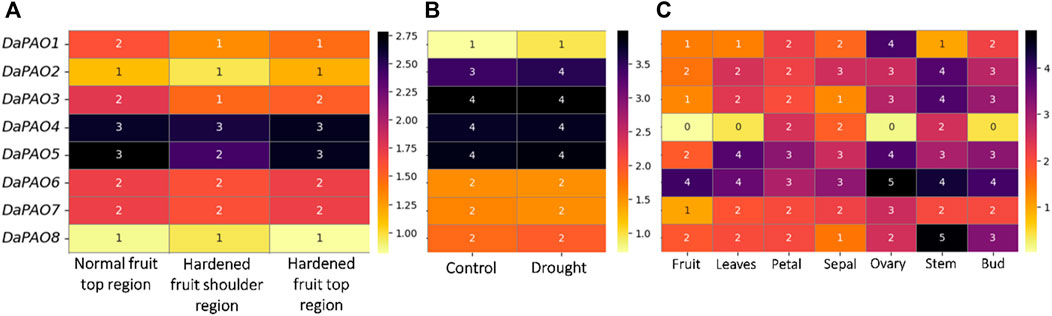
Figure 6. Heatmap showing the expression pattern of DaPAOs in (A) Disease condition where normal fruit region was compared with hardened fruit, (B) Drought stress condition, and (C) Different tissues, where yellow color shows downregulation and dark color shows upregulation.
3.8 3D structure prediction of DaPAO proteinsTo delve deeper into the structural and functional diversity, the 3D structures of the eight DaPAO proteins were modeled. These proteins exhibited a high degree of conservation in their 3D structures, with similar patterns of helices and turns. Additionally, all eight proteins shared a similar helical structure at both the C and N termini. This structural conservation implies that the DaPAO proteins may have similar functions. DaPAO7 has a slightly changed structure as compared to the other DaPAOs (Figure 7).
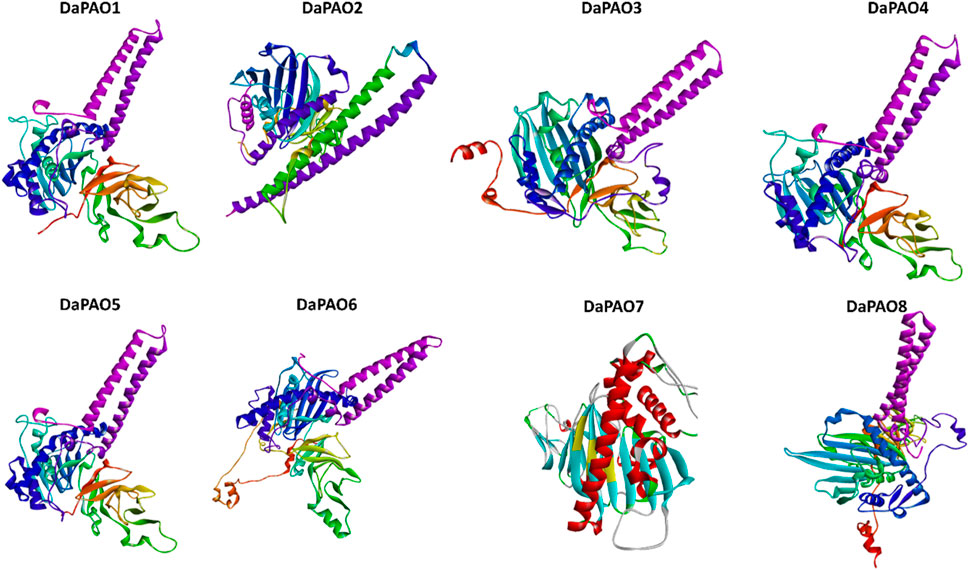
Figure 7. Predicted 3D structures of eight DaPAO proteins.
4 DiscussionPAO eventually creates Fluorescent Chl catabolites (FCCs) by opening the porphyrin macrocycle of pheophorbide a (Hörtensteiner, 2006). In the current study, a pangenome-wide study (Tahir ul Qamar et al., 2020) of this gene family has been carried out in eight Pyrus genomes. Here we identified 11 CuPAO genes, 8 ShPAO genes, 10 ZhPAO genes, 10 NiPAO genes, 10 YuPAO genes, 9 AnPAO genes, 5 BrPAO genes, and 8 DaPAO genes. All these members contained domains conserved in all PAO homologues. One essential component of leaf senescence is the phenotypic loss of chlorophyll, and the phenotype of mutants lacking in chlorophyll breakdown makes clear how important chlorophyll degradation is. For instance, the early cell death displayed by the maize lls1 mutant lacking in PAO eventually results in the death of the entire plant (Ougham et al., 2008; Das et al., 2018). Likewise, a phenotype of cell death is observed in rice PAO knockdown lines (Tang et al., 2011). Although the PAO/phyllobilin pathway of chlorophyll breakdown is important, it has only been studied in two monocot species thus far: rice, which is a cereal crop (Tang et al., 2011) and in a forage crop, ryegrass (Jespersen et al., 2016).
The phylogenetic tree revealed that Pyrus PAO proteins could be subdivided into four subfamilies, namely, Group A, B, C, and D. This division of clades is done on the basis of homologous relationships with members of inter- and intra-species. All the clades were shared by members of every genome used in this study. In pepper, the identified CaPAO also showed homology with the AtPAO members. PAO members from other genomes including Physcomitrella patens, Picea sitchensis, Selaginella moellendorffii, Nicotiana tobacum, Glycine max, Populus trichocarpa, and Solanum lycopersicum also showed similar homology pattern in phylogenetic tree (Tang et al., 2011).
By comparing Pyrus PAO the evolution of the members of this gene family was examined. It was found that almost all genes evolved through segmental duplication. Most of the Pyrus PAOs showed segmental duplication. However, at least one member from every single genome showed tandem duplication. In the promoter regions, cis-elements contribute to the stress responsiveness to environmental conditions a plant is exposed to. In Pyrus PAOs, cis-elements associated with light-related, hormone-related, stress-related, and development-related responsiveness were found which showed their involvement in abiotic as well as biotic stress responsiveness. The similar cis-elements have also been observed in PAO members from Arabidopsis which indicates their functional similarity and conservation across species (Aubry et al., 2018). Further, the functional prediction though PPI and GO analysis also showed the conservation and involvement of these genes in many metabolic reactions that take place in a leaf during senescence and loss of chlorophyll. The 3D structures necessary for performing proper functions was also found to be highly conserved among all members which also confirms the functional conservation among members identified from Pyrus genomes.
Pyrus plants are susceptible to a variety of biotic and abiotic challenges that can impair their growth and development, resulting in the loss of chlorophyll, cell death, and ultimately early senescence. The quality and productivity of crop plants would be impacted by each of these outcomes. Stresses of many kinds can have an impact on the expression and activity of PAO, an essential intermediary in the breakdown of chlorophyll. Apple leaves under drought had a significant upregulation in the relative expression of and PAO (Wang et al., 2013). According to microarray study, PAO overexpression in response to different stressors coincided with the decomposition of chlorophyll under these circumstances (Thomas et al., 2001). RNA-seq expression data analysis revealed that Pyrus DaPAO genes express differently in different tissues, disease condition, and the abiotic stresses (drought). The DaPAO6 and DaPAO8 were highly expressed in tissues while DaPAO1 exhibited down expression. In canola, BnPaO1 transcripts were only detectable in the early stages of seed development, but BnPaO2 expression was observable in seeds throughout seed development. When comparing BnPaO2 transcripts to BnPaO1 transcripts, the former displayed expression levels around 5.5 times greater, while the latter displayed levels of expression comparable to 21 DAP canola seeds at 8 to 10 DAP (Ho et al., 2006). Similarly, DaPAO4 and DaPAO5 showed an upregulation in normal and diseased fruit conditions. Moreover, in drought stress, four genes DaPAO2, DaPAO3, DaPAO4, and DaPAO5 showed an upregulation in gene expression while DaPAO1 showed a downregulation. Thus, these genes expressed significantly in abiotic and biotic stress conditions as well as in different developmental stages. Therefore, Pyrus PAO genes can be used in further research as this study has revealed their important role in stress responsiveness. In order to improve agricultural stress resilience, breeding and genetic engineering efforts involving the selection and integration of these genes may benefit greatly from the insights provided by these structural and functional perspectives.
5 ConclusionPAO is a crucial enzyme in the chlorophyll catabolism process. It ultimately generates the principal fluorescent chlorophyll catabolite and opens the porphyrin macrocycle. The present study provides a systematic as well as comparative analysis of PAO genes in eight economically important and nutritious Pyrus genomes. A total of 11 genes from Cuiguan genome (CuPAO), eight from Shanxi Duli (ShPAO), ten from Zhongai1 (ZhPAO), ten from Nijisseiki (NiPAO), ten from Yunhong No.1 (YuPAO), nine from d’Anjou (AnPAO), five from Bartlett v2.0 (BrPAO), and eight from Dangshansuli’ v.1.1 genome (DaPAO) were identified. We examined the physicochemical characteristics, evolutionary relationships, structural and functional conservation of these Pyrus PAO gene family members. Further, the cis-regulatory elements and expression analysis were also performed to analyze their expression in different stresses. These results are helpful to understand the roles of PAO genes in the various tissues, disease condition, and abiotic stress (drought). DaPAO5, DaPAO6, DaPAO7, and DaPAO8 are potential candidates which could help the pear confer tolerance against disease as well as drought stress. These genes can be subjected to genetic engineering research to create drought- and disease-tolerant crops, but more research is required to fully understand them. Additionally, our research will aid in the ongoing research on Pyrus’s PAOs functional roles to develop stress resistant Pyrus varieties.
Data availability statementThe original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributionsYM: Writing–original draft. JS: Writing–original draft. XZ: Writing–review and editing. MS: Writing–original draft. MT: Writing–review and editing. TL: Writing–review and editing.
FundingThe author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1396744/full#supplementary-material
ReferencesAubry, S., Fankhauser, N., Ovinnikov, S., Zienkiewicz, K., Feussner, I., Biology, M., et al. (2018). Pheophorbide a may regulate jasmonate signaling during dark-induced senescence. Plant Physiol. 182, 776–791. doi:10.1104/pp.19.01115
PubMed Abstract | CrossRef Full Text | Google Scholar
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic acids Res. 37, W202–W208. doi:10.1093/nar/gkp335
PubMed Abstract | CrossRef Full Text | Google Scholar
Chen, C., Xia, R., Chen, H., and He, Y. (2018). TBtools, a Toolkit for Biologists integrating various HTS-data handling tools with a user-friendly interface. arXiv. doi:10.1101/289660
CrossRef Full Text | Google Scholar
Chen, S., Sun, M., Xu, S., Xue, C., Wei, S., Zheng, P., et al. (2023). The pear genomics database (PGDB): a comprehensive multi-omics research platform for Pyrus spp. BMC Plant Biol. 23, 430. doi:10.1186/s12870-023-04406-5
PubMed Abstract | CrossRef Full Text | Google Scholar
Das, A., Christ, B., and Hörtensteiner, S. (2018). Characterization of the pheophorbide a oxygenase/phyllobilin pathway of chlorophyll breakdown in grasses. Planta 248, 875–892. doi:10.1007/s00425-018-2946-2
PubMed Abstract | CrossRef Full Text | Google Scholar
Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., et al. (2007). MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, 375–383. doi:10.1093/nar/gkm216
PubMed Abstract | CrossRef Full Text | Google Scholar
Fatima, K., Sadaqat, M., Azeem, F., Rao, M. J., Albekairi, N. A., Alshammari, A., et al. (2023). Integrated omics and machine learning-assisted pro fi ling of kinases from three peanut spp revealed their role in multiple stresses. Front. Genet. 2023, 1–17. doi:10.3389/fgene.2023.1252020
CrossRef Full Text | Google Scholar
Ferradini, N., Lancioni, H., Torricelli, R., Russi, L., Dalla Ragione, I., Cardinali, I., et al. (2017). Characterization and phylogenetic analysis of ancient Italian landraces of pear. Front. Plant Sci. 8, 751. doi:10.3389/fpls.2017.00751
PubMed Abstract | CrossRef Full Text | Google Scholar
Gasteiger, E., Hoogland, C., Gattiker, A., Wilkins, M. R., Appel, R. D., and Bairoch, A. (2005). “Protein identification and analysis tools on the ExPASy server,” in The proteomics protocols handbook (Spinger), 571–607.
CrossRef Full Text | Google Scholar
Goddard, T. D., Huang, C. C., Meng, E. C., Pettersen, E. F., Couch, G. S., Morris, J. H., et al. (2018). UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25. doi:10.1002/pro.3235
PubMed Abstract | CrossRef Full Text | Google Scholar
Gray, J., Wardzala, E., Yang, M., Reinbothe, S., Haller, S., and Pauli, F. (2004). A small family of LLS1-related non-heme oxygenases in plants with an origin amongst oxygenic photosynthesizers. Plant Mol. Biol. 54, 39–54. doi:10.1023/B:PLAN.0000028766.61559.4c
PubMed Abstract | CrossRef Full Text | Google Scholar
Hirashima, M., Tanaka, R., and Tanaka, A. (2009). Light-independent cell death induced by accumulation of pheophorbide a in Arabidopsis thaliana. Plant and Cell physiology 50, 719–729. doi:10.1093/pcp/pcp035
PubMed Abstract | CrossRef Full Text | Google Scholar
Ho, S., Chung, D. W., Pruz, A., and Ort, D. R. (2006). The role of pheophorbide a oxygenase expression and activity in the canola green seed problem. Plant Physiol. 142, 88–97. doi:10.1104/pp.106.084483
PubMed Abstract | CrossRef Full Text | Google Scholar
Hörtensteiner, S., and Kräutler, B. (2011). Chlorophyll breakdown in higher plants. Biochimica Biophysica Acta (BBA) - Bioenergetics 1807, 977–988. doi:10.1016/j.bbabio.2010.12.007
CrossRef Full Text | Google Scholar
Horton, P., Park, K.-J., Obayashi, T., Fujita, N., Harada, H., Adams-Collier, C. J., et al. (2007). WoLF PSORT: protein localization predictor. Nucleic acids Res. 35, W585–W587. doi:10.1093/nar/gkm259
PubMed Abstract | CrossRef Full Text | Google Scholar
Hu, B., Jin, J., Guo, A. Y., Zhang, H., Luo, J., and Gao, G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–1297. doi:10.1093/bioinformatics/btu817
PubMed Abstract | CrossRef Full Text | Google Scholar
Hunter, S., Apweiler, R., Attwood, T. K., Bairoch, A., Bateman, A., Binns, D., et al. (2009). InterPro: the integrative protein signature database. Nucleic acids Res. 37, D211–D215. doi:10.1093/nar/gkn785
PubMed Abstract | CrossRef Full Text | Google Scholar
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi:10.1038/s41587-019-0201-4
PubMed Abstract | CrossRef Full Text | Google Scholar
Le, D. T., Nishiyama, R., Watanabe, Y., Mochida, K., Yamaguchi-Shinozaki, K., Shinozaki, K., et al. (2011). Genome-wide expression profiling of soybean two-component system genes in soybean root and shoot tissues under dehydration stress. DNA Res. 18, 17–29. doi:10.1093/dnares/dsq032
PubMed Abstract | CrossRef Full Text | Google Scholar
Letunic, I., and Bork, P. (2007). Interactive Tree of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi:10.1093/bioinformatics/btl529
PubMed Abstract | CrossRef Full Text | Google Scholar
Li, J., Zhang, M., Li, X., Khan, A., Kumar, S., Allan, A. C., et al. (2022). Pear genetics: recent advances, new prospects, and a roadmap for the future. Hortic. Res. 9, 40. doi:10.1093/hr/uhab040
CrossRef Full Text | Google Scholar
Li, P., Ma, Y., Li, X., Zhang, L., Wang, Y., and Wang, N. (2006). Cloning and expressional characterization of soybean GmLls1 gene. Chin. Sci. Bull. 51, 1210–1218. doi:10.1007/s11434-006-1210-5
CrossRef Full Text | Google Scholar
Ma, N., Ma, X., Li, A., Cao, X., and Kong, L. (2012). Cloning and expression analysis of wheat pheophorbide a oxygenase gene TaPaO. Plant Mol. Biol. Report. 30, 1237–1245. doi:10.1007/s11105-012-0443-5
CrossRef Full Text | Google Scholar
Marchler-Bauer, A., Lu, S., Anderson, J. B., Chitsaz, F., Derbyshire, M. K., DeWeese-Scott, C., et al. (2011). CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, 225–229. doi:10.1093/nar/gkq1189
PubMed Abstract | CrossRef Full Text | Google Scholar
Mering, C. V., Huynen, M., Jaeggi, D., Schmidt, S., Bork, P., and Snel, B. (2003). STRING: a database of predicted functional associ
留言 (0)