
記住我






In adherence with the guidelines outlined in the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) framework for systematic review and meta-analysis reporting [27, 28], a systematic review was conducted. The review encompassed searches within the Medline, Cochrane, EMBASE, OVID, and PROSPERO databases, spanning the period from January 2000 to January 2024. The Systematic review was registered on the International prospective register of systematic reviews (CRD42023467540).
To effectively identify essential study concepts and facilitate the search procedure, we employed the Population, Interventions, Comparisons, Outcomes (PICO) framework in alignment with recommended practices [29]. The integration of the PICO framework within the field of health education is becoming increasingly essential, as it ensures comprehensive literature exploration and maintains relevance to the improvement of the health outcomes under investigation. Literature search strategy involved the utilization of Medical Subject Heading (MeSH) terms and free-text terms designed to refine the selection of relevant trials, specifically targeting the following items: “resistant hypertension”, and “aldosterone synthase inhibitors”, and “endothelin receptor antagonists”. As outcomes, we considered the “reduction of office (clinic) systolic/diastolic blood pressure” or “reduction of 24-h ambulatory systolic/diastolic blood pressure reduction”. Additionally, we conducted a review of the references cited in the articles identified during our search, to ensure the inclusion of comprehensive data.
For this study, stringent eligibility criteria were established to ensure the selection of relevant research material while maintaining scientific integrity. The study considered individuals of both sexes, aged 18 years or older, who had been diagnosed with resistant hypertension as eligible candidates for inclusion in the analysis. The primary focus of our analysis was centered on randomized clinical trials (RCTs) conducted specifically on patients undergoing optimal medical treatment for resistant hypertension. It is important to note that we specifically sought phase 2–3 RCTs, as these trials provide valuable insights on the effectiveness and tolerability of therapies when added to optimal medical treatment for resistant hypertension.
To ensure the precision and reliability of our research, certain types of articles were excluded. The excluded categories encompassed conference proceedings, abstracts, commentaries, reviews, observational studies, and case reports. The exclusion of these article types was imperative to ensure that the analysis primarily relied on phase 2–3 RCTs, thereby upholding the study’s scientific rigor, reducing bias, and ensuring the originality of the research findings.
Specifically, a total of 138,234 records was initially identified. After the removal of duplicates and initial screening, 127,879 records were excluded, as our inclusion criteria were limited to RCT exclusively and substantial portion of the initially retrieved articles did not align with the predefined research goals. Furthermore, 4277 additional records were excluded, as selection criteria were limited to studies conducted between 2011 and 2024. Several of the initially included articles, despite being filtered for RCT, were found to be non-pertinent to the purposes of our study. This discrepancy occurred as a result of the items utilized in the PICO format, which led to the selection of numerous trials unrelated to our research focus. Ultimately, our comprehensive review incorporated only five full-text articles.
A summary of the literature selection and screening process is presented in Fig. 2, adhering to the PRISMA guidelines. The key characteristics of selected randomized controlled clinical trials have been concisely summarized in Table 1.
Fig. 2
PRISMA 2020 flow diagram for updated systematic reviews, which included searches of databases and registers only.
Table 1 General characteristic of the study included in the systematic review3.2 Bias Risk Assessment and Quality of EvidenceWhen incorporating RCT into the analysis, it is essential to adopt the endorsed evaluation instrument, namely the updated iteration of the Cochrane tool, denoted as RoB 2 tool [30]. RoB 2 presents a structured framework for appraising the potential bias associated with a single outcome estimate derived from diverse types of randomized trials. RoB 2 is organized into distinct domains that encompass potential sources of bias impacting the outcome. The identification of these domains draws from a synthesis of empirical data and theoretical insights, enhancing its effectiveness as a bias assessment tool.
All the studies included in our analysis have consistently shown a low risk of bias, as indicated in Fig. 3 (online available), minimizing potential sources of systematic error or distortion in the results. A low risk of bias is a crucial aspect in ensuring the reliability and credibility of our findings, as it suggests that the data collected and analyzed in these studies are more likely to accurately represent the true relationships and effects under investigation. This strengthens the overall robustness and validity of our research.
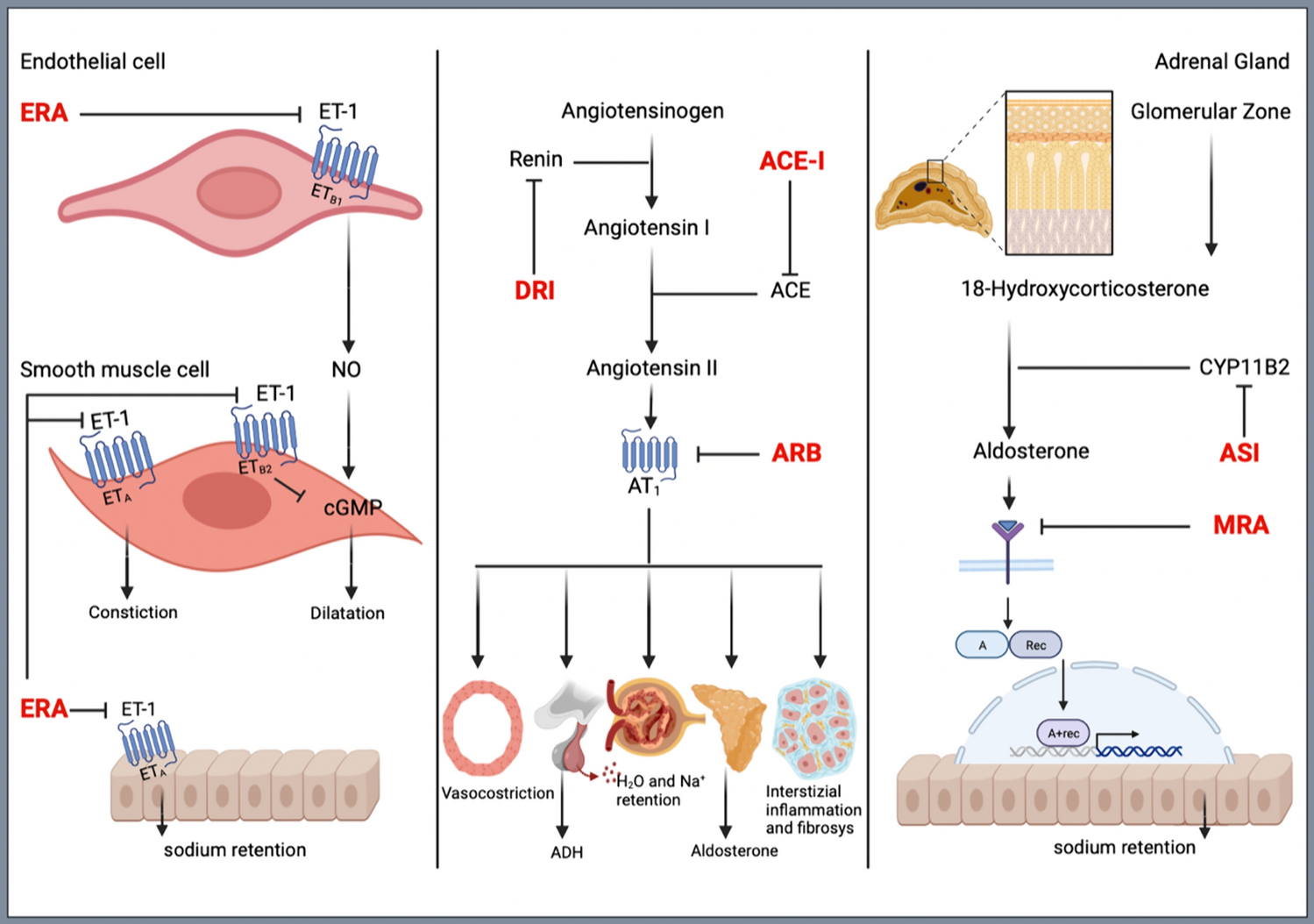
3.2.1 Aldosterone Synthase Inhibitors (ASI)Aldosterone is a key hormonal component of the renin-angiotensin-aldosterone system (RAAS). It is produced in the zona glomerulosa of the adrenal cortex and its production is enhanced by angiotensin II, high extracellular potassium concentration, and adreno- corticotropic hormone (ACTH). Aldosterone is involved in the regulation of fluid and electrolyte homeostasis via activation of the mineralocorticoid receptor, producing vasoconstriction of vascular smooth muscle and increased water and sodium retention by the kidneys at the distal tubule level. High levels of aldosterone can lead to hypokalemia, sodium reabsorption, and fluid retention, resulting in sustained BP elevation. The rate-limiting enzyme in the synthetic pathway from 18-hydroxycorticosterone to aldosterone is aldosterone synthase (also known as CYB11B2), as shown in Fig. 1.
According to guidelines, the preferred drugs for treating hyperaldosteronism and RHT are MRAs (spironolactone and eplerenone) [9, 10, 13]. More recently, ASI has been proposed as an alternative therapeutic strategy for reducing aldosterone production in the adrenal glands and aldosterone-related BP elevation [31, 32]. Some ASI agents have been developed and tested over the las few years. The results of clinic and 24-hour ambulatory BP reductions produced by these drugs at different dosages are summarised on Table 1.
A first agent in the ASI class was (LCI699, osilodrostat), which demonstrated to effectively correct hypokalemia in patients with primary aldosteronism in a proof-of-concept study [33] and to dose- dependently lower BP in patients with essential hypertension [34]. In this latter study, LCI699 was tested in a double-blind, randomized trial, performed in patients with primary hypertension [34]. After a 2-week of wash-out period and a 2-week single-blind placebo-controlled run-in period, eligible patients were randomly assigned to 1 of 6 treatment groups of double-blind treatment: LCI699 0.25 mg once daily, LCI699 0.5 mg once daily, LCI699 1.0 mg once daily, LCI699 0.5 mg twice daily, eplerenone 50 mg twice daily, or placebo for 8 weeks. Higher doses of LCI699 were not adopted, since preliminary studies indicated that these doses had the potential to inhibit 11--hydroxylase and to reduce cortisol synthesis [33]. The primary endpoint was changes in mean sitting diastolic BP compared to baseline values. Secondary endpoints included changes in mean sitting systolic BP and 24-h ambulatory systolic and diastolic BP changes from baseline.
From September 2008 to April 2009, 903 subjects were screened, 105 patients were excluded after the wash-out period, 274 during the single-blind period, 524 were randomly assigned to double-blind treatment, and 474 completed the 8-week double-blind period. At the end of the treatment period, LCI699 was associated with dose-dependent reductions in systolic BP levels of −9.7 mmHg, −8.7 mmHg, –12.6 mmHg, and –9.7 mmHg at the 0.25 mg, 0.5 mg, and 1.0 mg daily, and 0.5 mg twice daily, respectively, compared with placebo. However, eplerenone 50 mg twice per day produced lager and significant systolic BP reduction –13.8 mmHg) compared with placebo. With regard to the secondary endpoints, LCI699 1.0 mg daily (–7.1 mmHg; P=0.0012) and eplerenone 50 mg twice daily (–7.9 mmHg; P=0.0001) produced diastolic BP reductions compared with placebo (–2.6 mmHg); however, other doses of LCI699 did not provide significant changes in diastolic BP levels. Significant reductions in clinic systolic blood pressure were observed with all doses of LCI699 (P=0.005 or better) and eplerenone (P<0.0001). All doses of LCI699 significantly reduced systolic and diastolic 24-h ambulatory BP levels compared with placebo, though also in this case eplerenone 50 mg twice per day produced lager and significant systolic and diastolic 24-h ambulatory BP levels compared to placebo. Morning cortisol levels remained unchanged regardless of the dose of LCI699; however, ACTH stimulation of cortisol was suppressed in about 20% of patients receiving the higher doses.
In a subsequent randomized, double-blind, parallel-group, multicenter, dose-ranging study, the safety and efficacy of LCI699 was tested in RHT patients compared with either eplerenone or placebo [35]. After a 2-week single-blind placebo run-in, eligible patients were randomized (1:1:1:1:1) to receive placebo, eplerenone 50 mg twice daily or 1 of 3 doses of LCI699 (0.25 mg twice daily, 1 mg once daily, 0.5 mg then titrated to 1 mg twice daily after 4 weeks). All patients were treated for 8 weeks (any treatment discontinuation occurred) and a further follow-up safety visit was conducted 2 weeks after completing the study treatment. A total of 155 patients were randomized for the study and treated with study medication, and at the end of the treatment period, all LCI699 groups provided systolic and diastolic, clinic and 24-h ambulatory BP reductions, though none achieved statistical significance compared to placebo [35].
LY3045697 was the next ASI to be developed. This drug was tested in two small, placebo-controlled, crossover-designed clinical studies, which evaluated safety, pharmacodynamics, and pharmacokinetics under single and repeated dose conditions in otherwise healthy subjects, aged 18-65 years [36]. LY3045697 caused rapid dose and concentration-dependent reduction of SA levels, but it was not tested for BP lowering efficacy in humans [36].
Another drug in the ASI class is represented by CIN-107 (baxdrostat), which demonstrated high selectivity for the CYB11B2 enzyme inhibition, and lower affinity for 11b-hydroxylase compared to previous ASI agents, thus resulting in no relevant changes in plasma cortisol levels [37,38,39]. This drug was tested in preclinical and first-in-human clinical studies [40, 41] and more recently in phase 1, randomized, double-blind, multiple ascending dose study, which involved healthy volunteers [37]. This study aimed to assess safety, tolerability, pharmacokinetics, and pharmacodynamics of baxdrostat at different dosages (0.25, 0.5, 1, 2 mg once daily) for 10 days in subjects with a normal- or low-salt diet [37]. The low-salt diet cohorts were included to stimulate aldosterone production. The study demonstrated that baxdrostat produces a dose-dependent decrease in SA levels compared to baseline and placebo, while having no meaningful impact on serum cortisol levels [37]. The clinical effectiveness and safety of this drug in hypertension has been tested in a subsequent trial.
The BrigHTN trial was a multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-ranging trial, comparing the safety and efficacy of baxdrostat at different dosages with that of placebo [42]. The trial enrolled adult outpatients aged 18 years or more, with uncontrolled hypertension taking three or more antihypertensive drugs at maximally tolerated doses (one of which was a diuretic). Among these, about 45% were women, 70% were Caucasians, about half had obesity and 28% had diabetes. Baseline systolic/diastolic BP levels were 147/87 mmHg on average.
After a screening period of 8 weeks, and a subsequent 2-week, single-blind run-in period (during which medication adherence was assessed), eligible patients were randomly assigned to receive either 0.5 mg, 1 mg, or 2 mg of baxdrostat or placebo. The primary efficacy endpoint was the change in the mean seated systolic BP from baseline to the end of the 12-week treatment period in each baxdrostat group as compared with the placebo group. The secondary endpoints were the change from baseline in the mean seated diastolic BP and the percentage of patients with a seated BP of less than 130/80 mmHg at the end of the 12-week treatment period.
From July 2020 to June 2022 a total of 779 patients were screened, 360 entered in the run-in period and 275 patients were randomized. The main reasons for being excluded from the study were not reported. After the prespecified interim analysis, the trial was early stopped because the independent data monitoring committee concluded that the criteria for overwhelming efficacy were achieved. After 12 weeks of treatment, baxdrostat was associated with dose-dependent changes in systolic BP levels of −20.3 mmHg, −17.5±2.0 mmHg, and −12.1 mmHg at the 2-mg, 1-mg, and 0.5-mg doses, respectively. Compared to that observed in the placebo group (−9.4 mmHg), dosages of 2 mg daily (−11.0 mmHg; P<0.001) and 1 mg daily (−8.1 mmHg; P=0.003) induced significant systolic BP reductions, whereas the dose of 0.5 mg did not produce significantly different systolic BP changes. With regard to the secondary endpoint, baxdrostat 2 mg daily reduced diastolic BP by 14.3 mmHg, corresponding to −5.2 mmHg with that obtained in the placebo group.
Dose-dependent reductions in systolic BP levels were paralleled by reductions in serum aldosterone (SA) levels and compensatory increases in plasma renin activity (PRA), but not relevant effects on serum cortisol levels. Few adverse events were recorded (none serious).
Finally, lorundrostat is another agent in the ASI class to be tested in RHT patients. The Target-HTN trial was a multicenter, prospective, randomized, placebo-controlled, dose-ranging clinical trial, aimed at comparing the safety and efficacy of lorundrostat at different dosages with that of placebo [43]. The trial included adult outpatients aged 18 years or more, with uncontrolled hypertension taking two or more antihypertensive drugs at maximally tolerated doses. Among these, 60% were women, 36% were Afro-Americans and 48% were Hispanics; of note, about half had obesity, and 40% had diabetes. Baseline systolic/diastolic BP levels were 142/80 mmHg on average.
After 2–4 weeks of pre-screening and 2 weeks of placebo run-in period, eligible patients were stratified into two groups: cohort 1 enrolled participants with suppressed PRA ≤1.0 ng/mL/h and SA levels of 1.0 ng/dL, while cohort 2 enrolled participants with PRA greater than 1.0 ng/mL/h. Then, patients in cohort 1 were randomized to placebo or 1 of 5 dosages of lorundrostat (12.5 mg, 50 mg, or 100 mg once daily); in a subsequent group, patients were randomised to receive 12.5 mg or 25 mg of lorundrostat twice daily. Patients in the cohort 2 were randomized to receive placebo or 100 mg lorundrostat once daily. The primary efficacy end point was change in systolic attended office BP from baseline to the end of study week 8. Secondary efficacy endpoints included changes in diastolic attended office BP, and changes in 24-h ambulatory BP levels.
From July 2021 to June 2022, a total of 380 patients were screened and 200 were randomized (n=163 in cohort 1 and n=37 in cohort 2). The main reasons for being excluded from the study were inadequate PRA assessment. After 8 weeks of treatment in cohort 1, office systolic BP levels were reduced by 11.9, 13.7, and 5.6 mmHg with 100 mg, 50 mg, and 12.5 mg once daily of lorundrostat, respectively, and by 11.1 and 11.3 mmHg with 50 mg or 12.5 mg of lorundrostat twice daily, respectively. The corresponding differences from placebo in systolic BP change were –7.8, –9.6, –5.6 mmHg for once daily dosages and –7.0 and –7.2 mmHg for twice daily dosages. In cohort 2, lorudonstrat 100 mg reduced office systolic BP levels by 11.4 mmHg compared to placebo. With regard of secondary endpoint, lorundrostat 50-mg once-daily dose reduce diastolic attended office diastolic BP levels by 5.5mm Hg compared with placebo (P=0.02), whilst other doses did not provide statistically significant diastolic BP reductions compared with placebo. Also in this trial, few adverse events were recorded (none serious), mostly related to changes in serum potassium levels from baseline.
3.2.2 Endothelin A/Endothelin B Receptor AntagonistsEndothelin-1 (ET-1) is a vasoconstrictor peptide, produced in many different tissues, particularly in the endothelium of blood vessels. Endothelin-1 may act in a paracrine or autocrine way on blood vessels, by interacting with either ETA or ETB receptors on smooth muscle to stimulate contraction or on ETB receptors on endothelial cells to induce the release of vasorelaxants (nitric oxide and prostacyclin), thus contributing to regulate BP homeostasis [44]. Endothelin-1 production is increased in the presence of endothelial dysfunction and hypertension, and ERA has recently emerged as a novel target for high BP management and control. Indeed, preclinical studies demonstrated the efficacy of ERA in lowering BP [45], particularly in low-renin condition [46]. In these experimental models of hypertension, dual blockade of ETA/ETB receptors appeared to have a lower risk of fluid retention and vascular leakage than ETA-selective blockade. Aprocitentan is a potent, orally active, dual endothelin A/endothelin B (ETA/ETB) receptor antagonist, with long half-life (44 h).
The clinical effectiveness and safety of aprocitentan in hypertension has been tested in one dose-finding study [47] and in one phase 3 trial [48], both showing positive results in terms of systolic/diastolic BP reduction with good tolerability and safety profile [49]. It should be noted, however, that both these studies adopted unattended office systolic/diastolic BP measurements for efficacy and safety assessments; this may at least, in part, contribute to explain the high rate of screening failure reported in both studies [47, 48]. The results of clinic and 24-hour ambulatory BP reductions produced by this drug at different dosages are also reported on Table 1.
In a randomized, double-blind, multicenter, the efficacy and safety of aprocitentanat different dosages of 5, 10, 25, and 50 mg once-daily were evaluated in patients with grade 1–2 essential hypertension compared to that of either placebo or lisinopril 20 mg once daily [47]. After initial screening, patients entered in a single-blind, placebo run-in period for 4–6-weeks, then eligible patients were randomized to receive either placebo, or aprocitentan, or lisinopril at predefined doses. After 8 weeks of double-blind treatment, all patients entered a 2-week single-blind, placebo withdrawal period. Among 1,659 initially screened patients, 996 were enrolled in the placebo-run-in period, 490 were randomized and 430 patients completed the 8-week treatment period. At the end of the treatment period, significant dose-response changes in mean seated diastolic BP from baseline to week 8 were observed (P<0.001 for all 6 prespecified dose-response models) [47]. In those patients with valid ABPM at baseline and at week 8 (n=281), 10, 25, and 50 mg aprocitentan produced significant reductions in mean 24-hour systolic/diastolic BP levels from baseline by 3.99/4.04, 4.83/5.89, and 3.67/4.45 mmHg, respectively [47].
In the subsequent multicentre, double-blinded, randomised, parallel-group, phase 3 trial, the PRECISION trial [48], included adult outpatients with RHT, as defined by seated systolic BP of 140 mmHg or higher despite taking standardised background therapy with three antihypertensive drugs, including a diuretic. The study consisted of three sequential parts: (a) part 1 was a 4-week double-blind, randomised, and placebo-controlled period, during which patients received aprocitentan 12.5-25 mg, or placebo in a 1:1:1 ratio; (b) part 2 was a 32-week single-blind period, during which all patients received aprocitentan 25 mg; (c) part 3 was a 12-week double-blind, randomised, and placebo-controlled withdrawal period, during which patients were again randomised to either aprocitentan 25 mg or placebo in a 1:1 ratio; (d) part 4 was a 30-day safety follow-up period, during which all patients continued standardised background therapy. The primary endpoints were defined as changes from baseline to week 4 (part 1), and from withdrawal baseline (week 36) to week 40 (part 3), in mean sitting office systolic BP levels. Secondary endpoints included changes at week 4 and week 40 in mean sitting office diastolic BP and in 24-h ambulatory systolic and diastolic BP levels.
Overall, 1965 individuals were screened, 730 were randomised, 704 (96%) completed part 1; of these, 613 (87%) completed part 2 and, of these 577 (94%) completed part 3 of the study. In part 1, at 4 weeks, office systolic BP levels were reduced by –15,3 mmHg for aprocitentan 12,5 mg, –15,2 mmHg for aprocitentan 25 mg, and –11,5 mmHg for placebo; placebo-mediated differences were –3,8 mmHg (p=0·0042) and –3,7 mmHg (p=0·0046), respectively. Office diastolic BP levels also decreased with both aprocitentan doses compared with placebo (–3·9 and –4·5 mm Hg, respectively). During part 2, both systolic and diastolic office BP levels were maintained in patients previously receiving aprocitentan and decreased within the first 2 weeks of part 2 before stabilising in those previously receiving placebo. In part 3, after 4 weeks of withdrawal from the study drug, office systolic (5.8 mmHg, p<0·0001) and diastolic (5.2 mmHg, p<0·0001) BP significantly increased with placebo compared with aprocitentan.
Similar trends were observed in those patients who had valid 24-h ABPM. At the end of part 1 (week 4), aprocitentan decreased both the 24-h ambulatory systolic (–4.2 mmHg for the 12·5 mg dose and –5.9 mmHg for the 25 mg dose) and diastolic (–4.3 mmHg for the 12.5 mg dose and –5·8 mmHg, for the 25 mg dose) BP levels. At the end of part 3, after 4 weeks of withdrawal (week 40), both the 24-h ambulatory systolic and diastolic BP increased with placebo compared with aprocitentan (6.5 mmHg and 6.8 mmHg, respectively).
3.2.3 Non-steroidal Mineralocorticoid Receptor AntagonistsMineralocorticoid receptor antagonists (MRA), mostly spironolactone, have demonstrated to be effective in reducing BP in RHT patients [14]. These drugs are able to antagonize the binding of aldosterone to mineralocorticoid receptors, which are expressed in many tissues, including kidneys, thus reducing sodium retention, as schematically shown in Fig. 1. The use of these drugs, however, may be limited by the presence of renal impairment, chronic kidney disease (CKD) or dialysis. In addition, MRAs, particularly spironolactone, may be associated with adverse systemic effects, such as gynecomastia and sexual disorders. Current guidelines now recommend the use of MRAs for treating RHT patients only in the presence of an estimated glomerular filtration rate (eGFR) ≥45 mL/min/1.73m2 and a serum potassium concentration ≤4.5 mmol/L [9, 10, 13]. The most recent ESH 2023 hypertension guidelines [13] give preference to spironolactone as compared to other MRA for the treatment of RHT patients, mostly in view of the results of recent studies [14, 15, 50, 51] and meta-analyses [52,53,54].
Finerenone, a nonsteroidal MRA, has demonstrated to provide kidney protection in patients with advanced CKD and markedly elevated albuminuria included in the Finerenone in Reducing Kidney Failure and Disease Progression in Diabetic Kidney Disease (FIDELIO-DKD) trial [55], as well as in patients with type 2 diabetes and moderate-to-severe CKD with moderately elevated albuminuria or mild CKD with severely elevated albuminuria included in the Finerenone in Reducing Cardiovascular Mortality and Morbidity in Diabetic Kidney Disease (FIGARO-DKD) trial [56]. These trials [30, 31] were not planned to test efficacy of finerenone in RHT patients. Nevertheless, in a post-hoc analysis, RHT patients with advanced CKD and type 2 diabetes mellitus from FIDELITY (pooled population from the FIDELIO-DKD and FIGARO-DKD trials) was selected to evaluate BP lowering effects of this drug. Definition of RHT was based on the eligibility criteria adopted in the Spironolactone With Patiromer in the Treatment of Resistant Hypertension in Chronic Kidney Disease (AMBER) trial [57], a phase 2, multicenter, randomized, double-blind, placebo-controlled trial. In the FIDELITY-TRH post-hoc analysis [58], which included RHT patients with an eGFR of 25–45 mL/min/1.73 m2 and baseline serum potassium between 4.3 and 5.1 mmol/L. From baseline to week 17 office systolic BP was reduced −7.1 mmHg by finerenone and by −1.3 mmHg with placebo (between-group difference −5.74 mmHg, P<0.0001) [58]. In the AMBER cohort study, from baseline to week 12 office systolic BP was reduced by −11.7 mmHg by spironolactone + patiromer and −10.8 mmHg by spironolactone and placebo (between-group difference −1.0 mmHg; P=0,58) [58].
The mechanistic strategy underlying the potential use of finerenone in RHT cannot be identified with a novel therapeutic approach. However, the features of this compound, and namely the lack of the classical adverse effects of steroids, which are of clinical concern, and the demonstrated safety in a population with advanced CKD and diabetes, provide solid basis to further test the potential clinical advantages of finerenone in RHT.
留言 (0)