
記住我
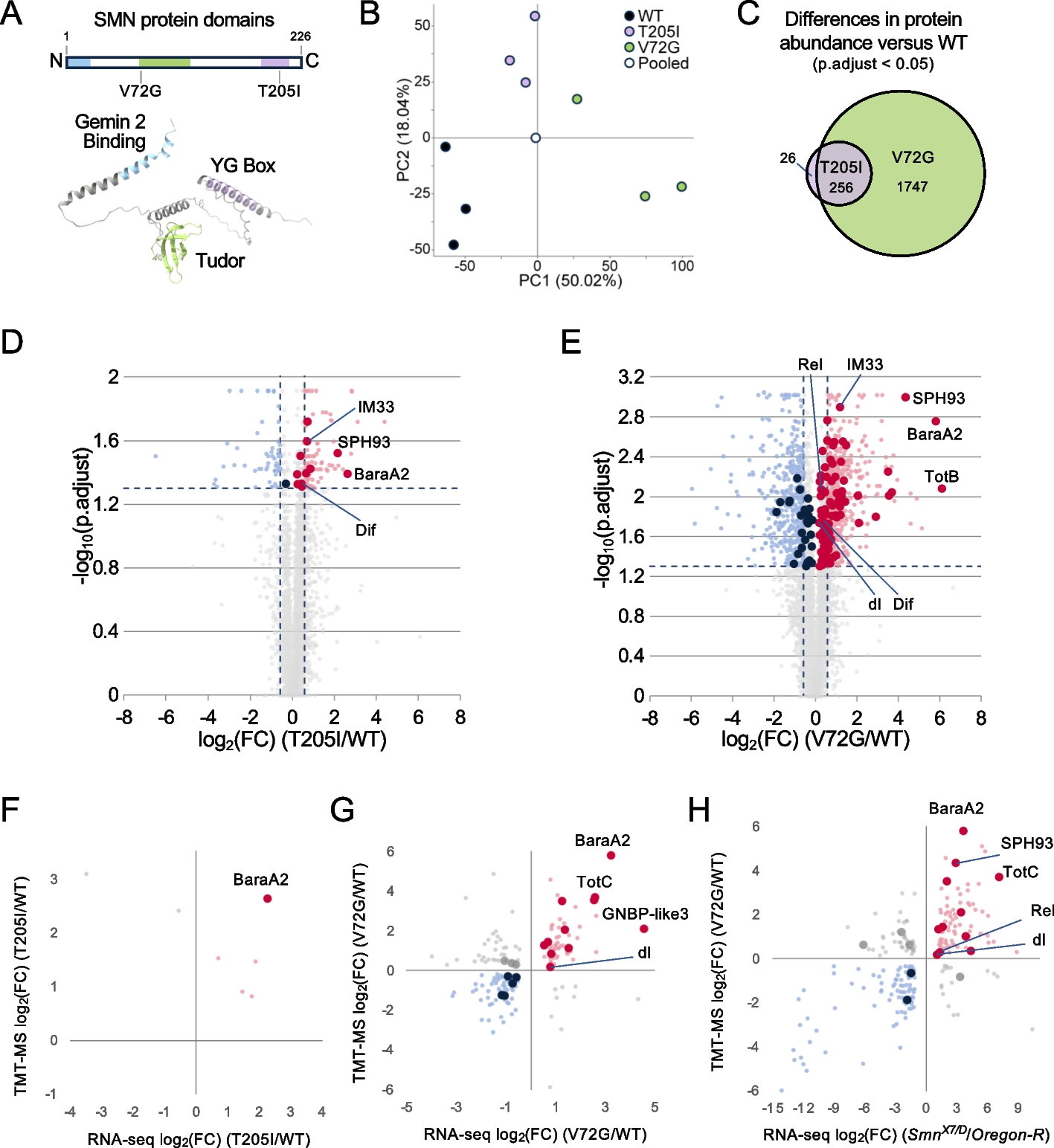

Using the PacBio Sequel II platform, we generated ~ 50 × long reads to obtain a high-quality genome for H. tibetana (2n = 2x = 24) (Fig. 1). The estimated genome size was 997.42 Mb based on Illumina short-read sequencing data (Table 1, Additional file 1: Fig. S1). Assembling the dataset with Hifiasm yielded a 957.16-Mb assembly, which is 95.9% the estimated genome size. There were 90 contigs generated from the assembly, giving the genome assembly a contig N50 of 36.40 Mb. Then, the PacBio sequence assembly was polished with 52.08 Gb (~ 52 ×) of Illumina sequencing reads to remove sequencing errors found in single-molecule sequence datasets. After that, a total of 100.78 Gb of Hi-C data were generated, which was integrated into the assembly (Fig. 2a). Consequently, 12 pseudochromosomes (scaffold N50 size, 88.98 Mb) of H. tibetana genome were successfully assembled, which contains nearly 92.99% (890.09 Mb) of the assembled sequences (Fig. 2b, Additional file 2: Table S1). The assembled sequences showed low GC content of 29.8%. Compared with the genome sequences of H. rhamnoides subsp. mongolia, our assembly (contigs N50 of 36.40 Mb) was obviously improved, which contained 30 scaffolds. Genome assembly coverage was evaluated by mapping high-coverage Illumina sequencing reads against the assembled genome. There was a 99.91% mapping rate for Illumina DNA reads and 97.03% for Illumina RNA reads.
Fig. 1
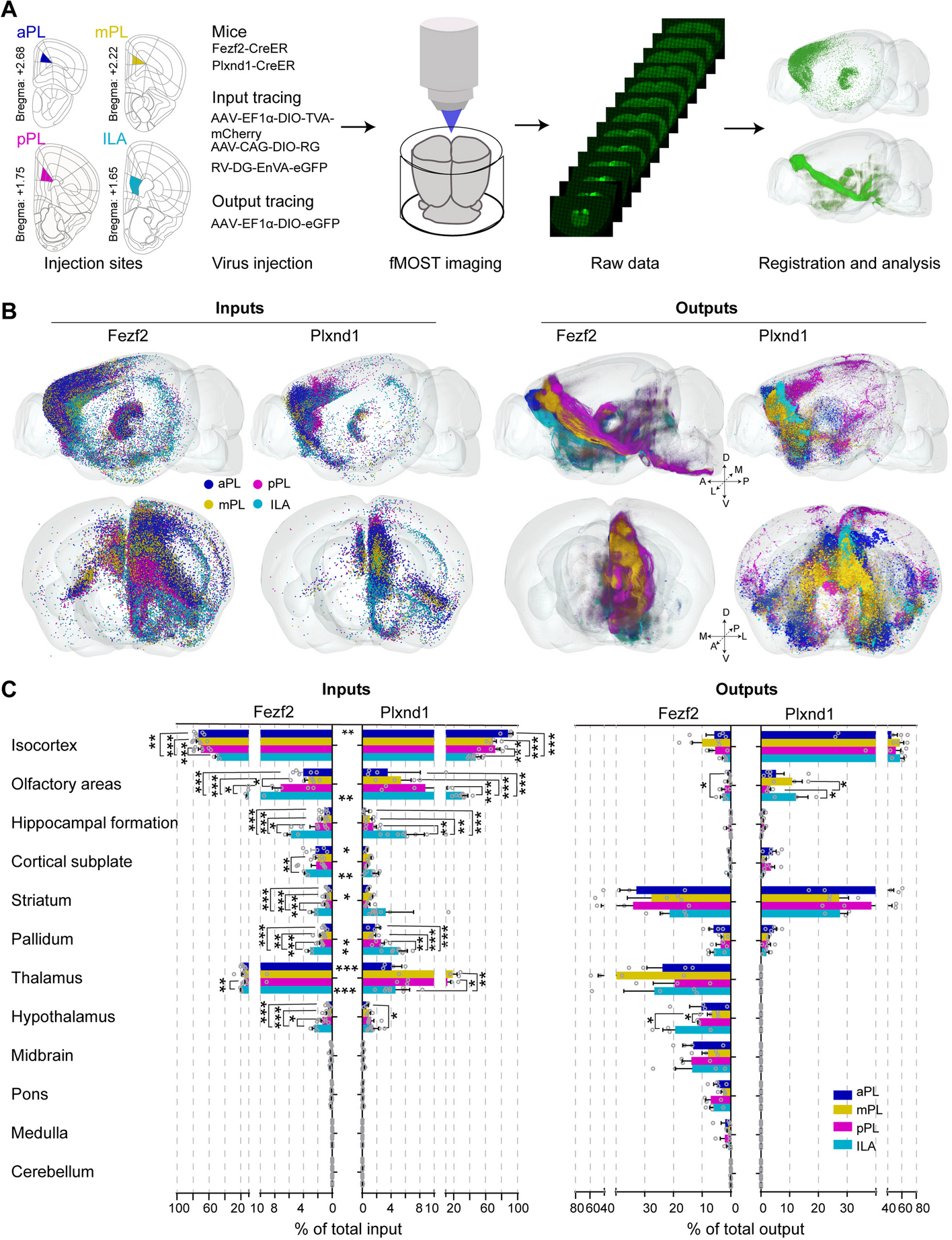
Sampling location and morphology of H. tibetana. a Sampling location of H. tibetana. b Photographs of H. tibetana trees. c Photographs of H. tibetana fruits
Table 1 Summary of the H. tibetana genomesFig. 2
Overview of the H. tibetana genome. a Genome-wide all-by-all interactions among all sea buckthorn chromosomes. b (A) Gene density and distribution (non-overlapping window size, 100 kb); (B–E) gene expression levels (Log2 FPKM) in sea buckhorn stem, root, leaves, fruit; (F) density of repeats (non-overlapping window size, 100 kb); (G) density of Copia-type transposons (non-overlapping window size, 100 kb); (H) density of Gypsy-type transposons (non-overlapping window size, 100 kb); (I) sea buckthorn pseudo-chromosomes. c Insertion time distribution of LTR retrotransposons
The assembled H. tibetana genome was 211 Mb larger than the reported genome of H. rhamnoides subsp. sinensis. In the H. tibetana genome, transposable elements (TEs) were more abundant than in the H. rhamnoides subsp. sinensis genome. It was found that TEs accounted for 605 Mb, or 66.01%, of the assembled H. tibetana genome, as compared to 335 Mb of TEs in the genome of H. rhamnoides subsp. sinensis (47.48%).
Gene prediction and functional annotationIn H. tibetana, 31,340 protein-coding genes were annotated based on de novo, homologous, and RNA sequencing predictions. The average length of genes and coding sequences were 4387 bp and 1176 bp, respectively (Additional file 2: Table S2). According to mapping results, 96.8% (30,345) of the predicted genes have a corresponding function (Additional file 1: Fig. S2). In addition, the sea buckthorn genome was predicted to contain 391 miRNAs, 941 tRNAs, 3459 rRNAs, and 4425 snRNAs (Additional file 1: Fig. S3, Additional file 2: Table S3). We assessed the quality and completeness of this assembled genome using two strategies. First, the H. tibetana genome contained 237 (95.56%) core eukaryotic genes by a core eukaryotic gene mapping approach (CEGMA) analysis. Then, a 98.2% (1614 genes) BUSCO recovery score demonstrated the high completeness of the assembly of the H. tibetana genome (Additional file 2: Table S4). These parameters of the H. tibetana genome are better than other published Elaeagnaceae Juss. genomes.
Through multiple de novo prediction procedures, we identified and classified repeat sequences within the repeat library using RepeatMasker (Additional file 2: Table S5). Approximately, 65.77% of the H. tibetana sequences were identified as repetitive elements, including retrotransposons (51.11%), DNA transposons (3.3%), and unclassified elements (14.76%). The most common type of retrotransposons (50.48%) were long-terminal repeats (LTRs), of which Copia and Gypsy constituted 17.55% and 17.26% of the genome, respectively. In H. tibetana, retrotransposons might be associated with genome expansion. Based on H. tibetana genome sequences, we estimated the insertion dates of LTR retrotransposons to explore its evolutionary dynamics. In H. tibetana, LTR retrotransposons are most abundant at ~ 1.0 Mya (Fig. 2c). As a result of LTR retrotransposons’ proliferation, H. tibetana has probably evolved to become more diverse, speciated, and adapted to high altitude conditions.
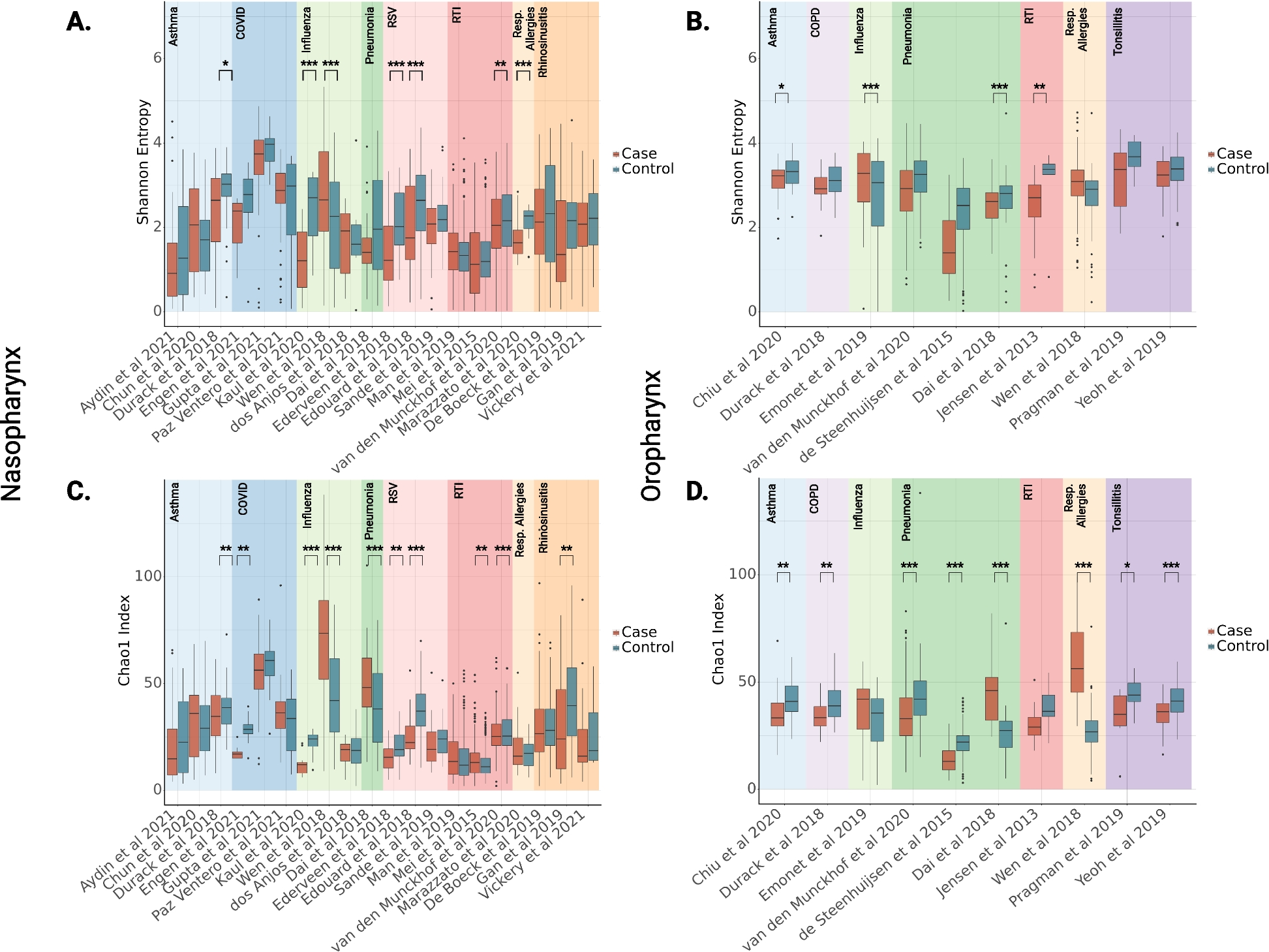
Evolution and gene family expansion/contraction analysisThe genome sequences of H. tibetana and another 10 plants (H. rhamnoides subsp. mongolia, H. rhamnoides subsp. sinensis, Populus trichocarpa, Ziziphus jujube, Vitis vinifera, Fragaria vesca, Arabidopsis thaliana, Malus domestica, Carica papaya, and Citrus sinensis) were analyzed to determine the expansion and contraction of genes in the genome of H. tibetana. We totally identified 25,729 orthogroups from 11 plant genomes, 8602 of which were common between all 11 plants (Fig. 3a, b). H. tibetana genome contained 281 species-specific orthogroups, containing 857 genes. These 857 genes were found to be significantly enriched in a number of biological processes, including protein processing in the endoplasmic reticulum, plant-pathogen interaction, ribosome biogenesis in eukaryotes, and ubiquitin-mediated proteolysis (Additional file 1: Fig. S3).
Fig. 3
Characterization of H. tibetana genome evolution and gene family expansion. a Distribution of gene numbers and family sizes in H. tibetana and 10 other representative species. b Venn diagram showing unique and shared gene families between genomes of H. tibetana and 4 close relatives. c A species tree on the basis of 1064 single-copy orthologues from H. tibetana and 10 other representative species. The black numbers represent the divergence times from present. Green and red indicate the numbers of significantly (p < 0.05) expanded and contracted gene families. d KEGG functional classification of contracted gene families. e KEGG functional classification of expanded gene families
The 1064 orthogroups with single-copy genes were identified from the 8602 common orthogroups. The single copy genes of the 11 plants were used to construct a phylogenetic tree (Fig. 3c). It is obvious that all 3 species of Hippophae L. (H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis) are clustered together. Also, in comparison with H. tibetana, H. rhamnoides subsp. mongolia and H. rhamnoides subsp. sinensis show the closest distance. According to the divergent analysis, H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis diverged from Ziziphus jujube at 79.7 Mya. The phylogenetic evolutionary relationship revealed that the H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis diverged at 7.6 Mya.
As a result of further analysis of gene clusters, numerous genes were found to have expanded or contracted in the 11 genomes. For the H. tibetana, a total of 430 expanded and 1426 contracted orthogroups were identified, respectively (Additional file 2: Table S6). Among 430 expanded orthogroups, 201 orthogroups with 1298 genes were significantly expanded (p < 0.01). According to the functional analysis of these 1298 genes (Fig. 3d), protein processing in the endoplasmic reticulum was the most enriched biological process. Among these expanded genes, a tryptophan decarboxylase (TDC) gene was found, which is involved in melatonin biosynthesis. Five CBL-interacting protein kinase (CIPK) genes were found, which play important roles in abiotic stress response. Two damage tolerance protein genes (rad31-1 and rad31-2) were UBA-related genes required for DNA damage tolerance. Five genes involved in flavonoid biosynthesis were found. Three calcium-binding protein CML10 genes regulate cold tolerance. We identified a gene (Chr08.46) that is involved in photooxidative stress responses and cell death induced by UV-C. In addition, we also discovered genes associated with disease resistance in plants, including 4 pathogenesis-related gene 1 and 2 fatty acid 2-hydroxylase 1-like (FAH2, Chr09.2289, Chr11.212) genes, produce reactive oxygen species after chitin treatment. A total of 6 genes encoding alpha-glucan endo-1,3-beta-glucosidase have been found, which is considered as a defense factor against fungal pathogens. Five dirigent protein-like (DIR) genes were identified, which are involved in the plant disease-resistance response.
There is often an association between the adaptive divergence of closely related species and the contraction in the size of particular gene families. A total of 1426 gene families were identified as significantly contracted genes in the H. tibetana genome compared to the genomes of 10 species (p < 0.05) (Additional file 2: Table S6). Based on KEGG enrichment analysis results, contracted genes were significantly enriched in plant secondary metabolite biosynthesis, cyanoamino acid metabolism, and starch and sucrose metabolism (Fig. 3e). Among these contracted genes, 104 genes were annotated as resistance-related genes, including 8 NBS-LRR genes, 13 LRR-RLK genes, 45 RLK genes, 2 RLP genes, 6 GLR genes, 13 GsSRK genes, 7 CRK genes, 3 GDSL esterase/lipase genes, and 7 laccase genes.
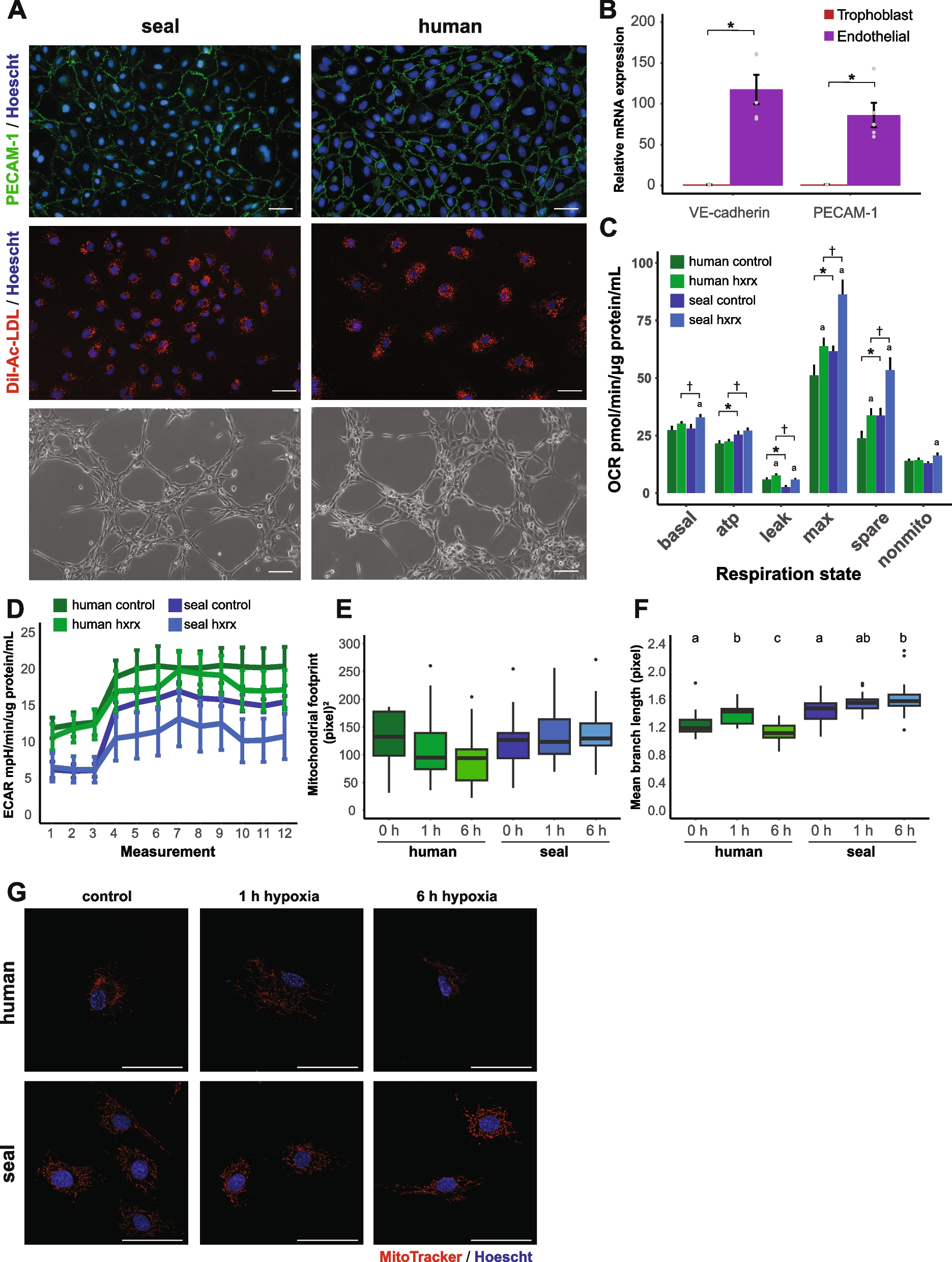
A whole-genome duplication (WGD) event is a key factor in the evolution of plants, animals, fungi, and other organisms. In order to investigate WGD in H. tibetana, we selected four species to perform a comparative genomics analysis. Based on the distributions of Ks of paralogous gene pairs in the H. tibetana genome, two recent clear peaks were found at 0.28 and 0.4 (Fig. 4a). There were two similar Ks peaks in the genomes of two closely related species, E. mollis and H. rhamnoides. Therefore, these two peaks might represent two closely WGD events in H. tibetana. These two WGD events were estimated to have occurred ~ 26 Mya and ~ 38 Mya. Phylogenetic evolutionary analysis indicated that these two WGD events occurred after Hippophae L. diverged from Jujuba.
Fig. 4
Identification of the WGDs in H. tibetana genome. a Ks distribution from orthologs between H. tibetana and other three species. b Gene Ontology (GO) functional classification of genes with WGDs. c KEGG functional classification of genes with WGDs
As a driving force for plant evolution, WGD has the potential to endow genes with new functions. GO enrichment analysis of genes associated with WGD events showed that these genes were significantly enriched into protein catabolic process, cellular protein catabolic process, and proteolysis involved in cellular protein catabolic process (Fig. 4b, c). Our KEGG enrichment analysis found that genes associated with WGD significantly enriched into plant hormone signal transduction, MAPK signaling pathways, fatty acid biosynthesis, and flavonoid biosynthesis (F3′H, CHS, C4H, CHI, DFR) (Fig. 4c). Thus, WGD events may related with high level of fatty acid and flavonoid content in sea buckthorn.
Candidate genes associated with high-altitude adaptation of the H. tibetanaGenes under positive selections were commonly associated with abiotic stresses, such as low temperature, low oxygen, reduced pathogen incidence, and strong UV radiation. The adaptive divergence of orthologs that show signs of positive selection usually occurs as a result of positive selection. Our study employed genomic sequences from H. tibetana and its 5 close species to conduct a positive selection analysis. We identified genes that show signs of positive selection from 1064 single-copy orthologs using the PAML 4 package’s branch-site model. The H. tibetana genome contained 212 genes possibly under positive selection (ω > 1, p < 0.05) (Additional file 2: Table S7). According to KEGG functional annotation, several of these genes are related to homologous recombination (RAD3-like DNA-binding helicase protein, DRD1 DEFECTIVE IN MERISTEM SILENCING 1, XRCC2 homolog of X-ray repair cross complementing 2, POLD3 DNA-directed DNA polymerase, RecQl3 DEAD/DEAH box RNA helicase family protein), Mismatch repair (5′-3′ exonuclease family protein, POLD3 DNA-directed DNA polymerase), ubiquinone and other terpenoid-quinone biosyntheses, base excision repair (poly ADP-ribose polymerase 3, POLD3 DNA-directed DNA polymerase), and plant-pathogen interaction (MKK6 MAP kinase kinase 6, calcium-binding EF-hand family protein). We identified genes that encode the RAD3-like DNA-binding helicase protein, DNA-directed DNA polymerase (POLD3), homolog of X-ray repair cross complementing 2 (XRCC2), DEAD/DEAH box RNA helicase family protein, 5′-3′ exonuclease family protein, and poly ADP-ribose polymerase 3, providing evidence that adaptive evolution in plants depends on positive selection of genes. In addition, two plant-pathogen interaction genes (MKK6 MAP kinase kinase 6 and calcium-binding EF-hand family protein) were also under positive selection, indicating that positive selection of genes may have a close correlation with plant-pathogen interaction in TQP.
Genome comparison between H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensisA variety of genetic variations, such as insertions and deletions, cause genetic diversity and phenotypic variation. Compared to H. rhamnoides subsp. mongolia and H. rhamnoides subsp. sinensis, H. tibetana exhibited obvious phenotypic differences, which allowed us to analyze the genomic variations between these three species. Full-genome alignment results revealed 237 syntenic blocks of H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis. These syntenic blocks contained 21,623 and 21,905 conserved genes covering 68.99% and 69.89% of the H. tibetana genome, respectively (Fig. 5a). In H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis, there are many collinear blocks, suggesting there is a high level of gene collinearity between the genomes (Fig. 5a). We identified 9,932,195 SNPs and 11,120,000 SNPs in H. tibetana genome compared with H. rhamnoides subsp. mongolia and H. rhamnoides subsp. sinensis genome (Fig. 5b, c). In addition, we identified 58,645 inversions, 13,967 deletion, and 11,305 insertion in H. tibetana genome compared with H. rhamnoides subsp. mongolia genome (Fig. 5b). Less deletion and insertion were found between H. tibetana and H. rhamnoides subsp. sinensis genome (Fig. 5b). Following this, we conducted structural variation analysis in the exon, promoter (2 kb up-5′UTR) and downstream (1 kb down-3′UTR) regions of gene in the H. tibetana genome. Compared with H. rhamnoides subsp. mongolia genome, we identified 2900 SV in the exon regions, 11,146 SV in the promoter regions, and 8616 SV in the downstream regions (Fig. 5b). Less SVs were found between H. tibetana and H. rhamnoides subsp. sinensis genome (Fig. 5c, d). H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis genomes show great variation possibly explaining their phenotypic differences.
Fig. 5
The comparison of H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis genomes. a Syntenic blocks share between the H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis. b The numbers of SNPs, delection, insertion, and inversion in the H. tibetana genome compared with H. rhamnoides subsp. mongolia and H. rhamnoides subsp. sinensis. genome. c The distribution of structural variation (SV) in the exon, intron, UTR, promoter (2 kb up-5′UTR), and downstream (1 kb down-3′UTR) regions of genes in the H. tibetana genome compared with H. rhamnoides subsp. mongolia genome. d The distribution of structural variation (SV) in the exon, intron, promoter (2 kb up-5′UTR), and downstream (1 kb down-3′UTR) regions of genes in the H. tibetana genome compared with H. rhamnoides subsp. sinensis genome
Investigation of genes involved in the flavonoid biosynthetic pathwayPast researches reported the contents of phenylpropanoids and flavonoids were associated with the tolerance to UV light of Tibetan peach, Arabidopsis, and rice in TPQ [6, 21]. In plants, flavonoid biosynthetic genes have been identified and characterized (Fig. 6a). In the genome of H. tibetana, 49 homologs of genes associated with flavonoid biosynthesis were identified (Additional file 2: Table S8). Gene cluster analysis results showed that all these genes were divided into 3 groups (Fig. 6b). Most genes (44 genes) were expressed in all 4 different tissues, according to gene expression analysis. The root had higher expression levels for 17 genes involved in flavonoid biosynthesis than other tissues (Fig. 6b). In the fruit, 13 genes were more highly expressed in the flavonoid biosynthesis pathway (Fig. 6b). For example, the fruit exhibited extremely high expression levels of a F3H gene (HtChr08.706) (FPKM > 2571). In contrast, 2 C4H (HtChr04.654 and HtChr07.1953) genes have higher expression levels in the roots, leaves, and stem tissues, indicating the 2 genes might be related with the more flavonoid content in the roots, leaves, and stem of H. tibetana (Fig. 6c, d).
Fig. 6
Genes associated with flavonoid biosynthesis in sea buckthorn. a Identification of structural flavonoid biosynthesis genes in H. tibetana. b Expression level of genes involved in the flavonoid biosynthesis pathway was detected in four different tissues of H. tibetana. The red colors indicated high expression levels. The blue colors indicated low expression levels. c the total flavonoid content of four different tissues of H. tibetana. d Maximum-likelihood trees of C4H family genes that were constructed using the amino acid sequences with 1000 bootstrap repeats. e Induction process of sea buckthorn hairy roots. CK, empty vector; OE-HtC4H1, overexpression lines (K599-1302-HtC4H1); OE-HtC4H2, overexpression lines (K599-1302-HtC4H2). f Relative expression levels of HtC4H1 and HtC4H2 in normal and transgenic hairy roots determined by qPCR. **p < 0.01, *p < 0.05. g Determination of flavonoids in CK and OE of hairy roots
Genomics and RNA sequencing provide valuable information for metabolic engineering that can enhance flavonoid content (Additional file 1: Figs. S4 and S5). The phenolic biosynthesis pathway starts with C4H, which is important for flavonoid accumulation in many plants. The flavonoid content of H. tibetana hairy roots was increased by overexpressing two C4H genes (HtC4H1 and HtC4H2) (Fig. 6e–g). We found that transgenic hairy roots showed higher flavonoid content compared with normal hairy roots (Fig. 6g). Hence, sea buckthorn flavonoids can be produced by cultivating hairy roots on a large scale.
留言 (0)