
記住我


HEK293T cells (CRL-3216, American Type Culture Collection) were transfected with the HIV-1 pro-viral DNA construct pNL4-3 by polyethylenimine as described85. The cell culture medium was exchanged with fresh medium at 6 h post-transfection and the supernatant was collected at 72 h for virion RNA extraction and p24 particle release measurement by the HIV-1 p24 enzyme-linked immunosorbent assay (ELISA) kit (Abcam). Total RNA from the cells was extracted by TRI reagent (Sigma-Aldrich) following the manufacturer’s instructions. Total viral particles were concentrated and centrifuged for 1 h 40 min at 28,000g at 4 °C with a 10% sucrose gradient. After discarding the supernatant, the pellet was resolved using 160 μl of 1× Hanks’ balanced salt solution, followed by DNase treatment for 30 min at 37 °C. The sample was incubated in 1 ml TRIzol for 5 min, followed by the addition of 200 μl chloroform and shaking for 30 s. The tube was then centrifuged for 10 min at 12,000g at 4 °C. The clear upper aqueous layer, which contains RNA, was transferred to a new 1.5 ml tube and 0.7 ml of isopropanol was added. After 10 min incubation at room temperature, the tube was centrifuged for 10 min at 12,000g at 4 °C. The supernatant was discarded, and then the pellets were resuspended in 1 ml of 70% ethanol, followed by another centrifugation for 5 min at 7,500g. The RNA pellet was air-dried in the hood and then resuspended in 30 μl of diethyl pyrocarbonate-treated water.
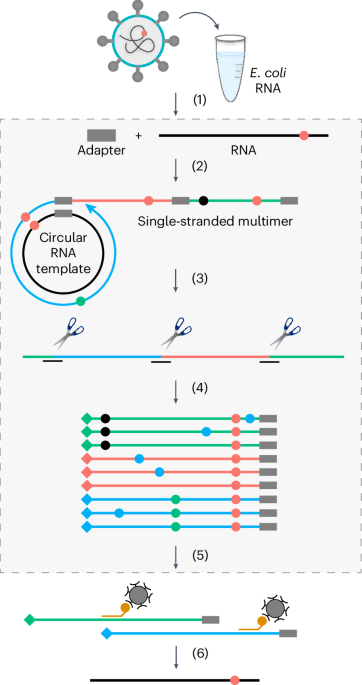
Nanopore DRS of full-length HIV-1 RNAFor HIV-1 RNA DRS, 1 μg of virion RNA or 10 μg of total cellular RNA in 9 μl was used for DRS library preparation following the manufacturer’s protocol (the Oxford Nanopore DRS, SQK-RNA002) with a modification of the RT step. Mixtures of 111 HIV-1 sequence-specific DNA oligomers (Integrated DNA Technologies) at a copy number ratio of 1:30 (HIV-1 RNA:each oligomer) were annealed to the RNA at 65 °C for 5 min and proceeded to the RTA ligation step. The DNA oligomers used are listed in Supplementary Table 9. The library was run on FLO-MIN106D flow cell for 48 h on a MinION device (Oxford Nanopore Technologies). IVT RNA fragments were sequenced the same way except 4 μg RNA was used for the input.
DRS data preprocessingMinKNOW GUI (v.3 or later; Nanopore Technology) was used for sequencing data collection. Multi-fast5 reads were base-called by guppy (v.3.2.8 or higher) using the fast base calling option. The base-called multi-fast5 reads were then converted to single-read fast5s using the Oxford Nanopore Technologies application programming interface, ont_fast5 (v.3.3.0). Fastqs were aligned to the HIV-1 genome reference sequence AF324493.2 from the National Center for Biotechnology Information (NCBI) or the human reference sequence (human genome assembly GRCh38.p13 for Extended Data Fig. 7a) with the options ‘-ax map-ont’ using minimap2 (v.2.24). Unmapped reads were discarded using SAMtools (v.1.6). For in-depth analysis of the 3′-end region, short reads (read length <2,000) were filtered out using NanoFilt (v.2.7.1). The sequence read length was extracted by aligning sequences against the HIV-1 coding sequences retrieved from HIV-1 genome reference sequence AF324493.2 from NCBI using minimap2 (v.2.24), retaining multiple secondary alignments (parameters -p 0 -N 10) and counting the number of unique read IDs among mapped alignments. Reads were required to be over 8,000 nucleotides to be classified as full-length HIV-1 RNA. Sequencing read coverage depth was calculated using bedtools genomcov of v.2.25.0 and visualized for 1 nt binning size in the plots.
DRS-mediated detection of RNA modificationsNanopore DRS can detect various types of chemical modification based on the DRS electrical signals (raw current intensity and dwell time) and/or the detection of modification-induced base-calling errors. For a whole-genome scale analysis of chemical modifications on HIV-1 RNA, we used Tombo (v1.5.1, based on current intensity differences32; section 1.2), Eligos2 (v.2.0.0, based on error rates33; section 1.3), Nanocompore (v.1.0.4, based on both current intensity and dwell time differences34; section 1.4) and Xpore (v.2.1, based on current intensity differences at the individual read level35; section 1.4). We also used Nanom6A40 (v.2.0) and m6Anet49 (v.1.1.1) for a read-level detection of m6As (section 1.5). We then cross-compared the results of Tombo, Eligos2 and Nanocompore to identify the most likely candidates of RNA modification sites (section 1.6). Default options were used for all software used in this study, except where noted.
Preparation of IVT RNA controlsWe generated three types of HIV-1 IVT datasets: (1) full-length (with the identical nucleotide sequence to NL4.3 RNA from the nucleotide position 1 to 1,973 with a poly(A) tail at the 3′ end), (2) half-length (F1 fragment from 1 to 4,587 and F2 fragment from 4,588 to 9,173) and (3) short IVT (7 fragments of 1 to 2 kb covering the whole genome) (Supplementary Fig. 1). The full-length IVT RNA reads were used for the whole-genome scale comparison of RNA modification sites (Fig. 1). The half-length IVT datasets were used to train m6Arp models (see the ‘Machine learning: determining m6A modifications per-read per-position basis’ section). We found the short IVT RNA sets are not suitable for the whole-genome scale analysis due to unreliable modification signals at the ends of RNA reads (see the ‘Determining the signal instability at the first and the last 40 nucleotides of DRS reads’ section below).
Tombo analysisTombo software uses raw DRS current intensity data to identify modified bases. DRS raw signals can distinguish canonical and non-canonical bases within the read head of the pore protein, but they also reflect various contexts surrounding the read head, including local RNA structures and neighbouring nucleotide sequences interacting with pore or motor proteins32,86. Given the high levels of noise in Tombo de novo analysis, we employed a two-step noise-reduction procedure to refine modification signals as follows:
Step 1 Tombo-MSCTombo de novo analysis, calculating current intensity deviations at each site using the ‘expected canonical signal levels’, generated highly noisy modification signals (Supplementary Fig. 2). To reduce the noise, we employed Tombo model–sample–compare (MSC) for a more accurate modification detection using HIV-1 IVT RNA reads, which adjust the ‘expected canonical signal levels’ for HIV-1 RNA-specific analysis (using the ‘—sample-only-estimates’ option). As expected, Tombo’s Tombo-MSC generated HIV-1-specific per-read P values for modification sites and population-level ‘estimated fractions of significantly modified reads’ (dampened_fraction or d values; Extended Data Fig. 2a), which successfully dampened the majority of noise signals observed in Tombo de novo analysis (Supplementary Fig. 2). For a full-length comparison of modification sites, we used a total of 5,411 of full-length IVT RNA reads as a canonical control for Tombo-MSC (Fig. 1 and Extended Data Fig. 2a). For an analysis using the half-length IVT data, we used 26,000 reads of the first half (F1) and 28,100 reads of the second half IVTs (F2), which resulted in a similar read coverage (~25,000) for the whole genome (Extended Data Fig. 2b). Tombo-MSC using 25,000 reads of IVT RNAs generated per-read P values that are highly reproducible, showing r2 > 0.999 when compared with the P values generated with 50,000 IVT reads (Extended Data Fig. 2c) and r2 = 0.8852 ± 0.0244 for four different WT HIV-1 virion RNA datasets prepared independently (Extended Data Fig. 2d).
Step 2 removal of the baseline noiseAlthough significantly reduced compared with Tombo ‘de novo’ analysis, Tombo-MSC analysis showed a considerable level of the baseline d value noise in our control analysis using 1,450 reads of full-length IVT reads (IVT subreads that were not used as a canonical control; Supplementary Table 8). The d values of IVT subreads coincided with most of d values of native HIV-1 RNA (Supplementary Fig. 3). A subtraction of the baseline noise substantially refined the modification signals of virion RNA (Supplementary Fig. 3).
A similar two-step noise-reduction procedure using IVT subreads was applied to all pretrained detection tools used in this study, including ELIGOS2 (Supplementary Fig. 5), nanom6A and m6Anet (Supplementary Fig. 6). DRS analysis 1.3. Eligos2 analysis: Eligos2 identifies the position of modification based on the differences in ‘error at specific base’33 between the native HIV-1 RNA and IVT RNA. Similar to Tombo-MSC analysis, we generated odd ratios of error at specific base at each position for both native RNA and IVT subread data using 5,411 reads of full-length IVTs as a canonical control (Supplementary Fig. 5). The modification signals (odd ratios) of native RNA were refined by subtracting the baseline noise (1,450 IVT subreads).
Dwell time was extracted for per-read and per-position levels using the Tombo Python application programming interface. For Tombo-MSC ‘d values’ (‘estimated fraction of significantly modified reads’), see Extended Data Fig. 2a.
Determining the signal instability at the first and the last 40 nucleotides of DRS readsNanopore DRS fails to read the 5′ end (the first 10–12 nucleotides) of RNAs due to the instability of the ends of RNA during the DRS runs27,28,87. In our data analysis, we also found that the electric signals of both ends of DRS reads (although successfully read by DRS) can still be unreliable (Supplementary Fig. 4a). To clearly address this, we evaluated the stability of local DRS signals by comparing long (F1 and F2) and short IVT RNA (F3–F9) reads with identical nucleotide sequences (Supplementary Fig. 4b). A systemic evaluation of DRS signals per position from each end (5′ or 3′) of the reads showed that DRS signals of the first 10–40 nucleotides and the last 10–40 nucleotides are not reliable, showing significant difference compared with the identical sequences in the middle of long RNA reads (P < 0.01; Mann–Whitney U tests comparing every pair of 10 base bins between long and short IVT reads; Supplementary Fig. 4b(ii)). For an accurate detection of RNA modifications, we excluded the DRS data of the first and the last 40 nucleotides.
Nanocompore and xPore analysesTo detect modified nucleotides, we also used Nanocompore (evaluating current intensity and dwell-time differences)34 and xPore (evaluating current intensity differences within a read)35 comparing raw DRS signals between native HIV-1 RNA and IVT RNA (Fig. 1 and Extended Data Fig. 3). Tools that compare raw DRS signals of two comparing samples, including Nanocompore, xPore and Tombo-Level Sample Compare (Tombo-LSC), do not require the noise removal using IVT subreads.
Nanom6A and m6Anet analysesFor a read-level detection of m6As, we employed Nanom6A40 and m6Anet49, which are pretrained tools. Similar to the Tombo-MSC and Eligos2 analyses described above, we generated m6A modification ratios for both native RNA and IVT subread data and performed a baseline noise removal to refine the m6A data (Supplementary Fig. 6).
Determining common modification sites among Tombo, Eligos2 and Nanocompore resultsThe sensitivities of DRS signal features, including current intensities, dwell time and base-calling error rates, can vary depending on the types and positions of the modifications36. Among all the tested in our study, Tombo, Eligos2 and Nanocompore generated the most reproducible results (Extended Data Fig. 3). Given approximately 80–200 modifications of various kinds may exist per genome based on previous mass spectrometry studies5,12, we selected and compared the 149 strongest peaks of modification signals from Tombo-MSC (d value >0.05), 167 from Eligos2 (odd ratios >2.4) and 156 from Nanocompore results (logit log-odd-ratio (LOR) score >0.73) (Supplementary Fig. 7a). To determine most probable modification sites among the myriad of modification signals in these analyses, we cross-compared these datasets and identified common peaks of these analyses (Supplementary Fig. 7b–c).
The probability that 14 out of 25 common sites coincide with DRACH sitesWe found that 14 out of 25 common modification sites (from Tombo and Eligos 2 analysis) on or one base away from the centre of the DRACH site (m6A sequence motifs; Supplementary Fig. 7c). To test whether this frequency is simply a random event (null hypothesis), we generated 25 random sites on the HIV-1 genome and calculated their chances to locate on or one base away from the centre of the DRACH sites on the HIV-1 genome. To be considered ‘on a DRACH’ (or ‘success’), a random event must occur within six bases (either upstream or downstream) of the centre of a DRACH site. The sixth base was chosen given the five-base resolution of Nanopore signals and a one-base margin as defined in Supplementary Fig. 7b(iii). The probability of a random site to be ‘on a DRACH’ (success) is approximately 0.313 = 2,867/9,173: (2,867; number of nucleotides within 6 bases of the centre of DRACH sites on the HIV-1 genome)/(9,173; number of nucleotides of the HIV-1 genome). Given that the probability distribution of 25 random events (from 0 success to 25 successes) is a binomial distribution, we calculated that the probability to have 14 or higher successes is approximately 0.00893 (see the probability distribution in Fig. 1d). This is sufficiently low to reject the null hypothesis; the chance that 14 out of 25 common sites coincide with DRACH sites is highly unlikely to be random.
The probabilities that m5C, ac4C and m6A-reader-binding sites to coincide with the 25 common sites were also calculated as described above using the m5C- and ac4C-detected areas and m6A-reader binding areas (Supplementary Table 4).
In vitro demethylation of m6A on HIV-1 RNARecombinant ALKBH5 (active motif) was used for in vitro treatment of HIV-1 virion RNA as described88. The reaction mixture contained KCl (100 mM), MgCl2 (2 mM), RnaseOUT (Invitrogen), l-ascorbic acid (2 mM), α-ketoglutarate (300 μM), (NH4)2Fe(SO4)2·6H2O (150 μM) and 50 mM of HEPES buffer (pH 6.5). The mixture was incubated for 1.5 h at room temperature and then stopped by the addition of 5 mM EDTA.
m6A dot immunoblottingThe extracted HIV-1 virion RNA was directly used for dot-blot assays, as previously described85. Briefly, 50 ng of virion RNA, diluted in 1 mM EDTA (total 100 μl), were mixed with 60 μl of 2× saline sodium citrate (SSC) buffer (Invitrogen) and 40 μl of 37% formaldehyde (Invitrogen). The mixture was incubated at 65 °C for 30 min. The nitrocellulose membrane (Bio-Rad) and nylon membrane (Roche) were both soaked with 10× SSC for 5 min before loading the RNA samples. Samples were loaded equally on nitrocellulose and nylon membrane followed by washing with 10× SSC buffer. The nylon membrane was washed with 1× TBST (25 mM Tris, 0.15 M NaCl and 0.05% Tween 20) and stained with methylene blue while shaking for 2–5 s and washed with ddH2O. The nitrocellulose membrane was UV cross-linked and then blocked with 5% milk in 1× TBST 1 h. m6A levels were detected by using an m6A-specific antibody (Abcam, cat. no. ab208577,1:1,000). Images were analysed by ImageJ software (v.1.53), and the relative RNA m6A levels were normalized to methylene blue staining.
LC–MS/MS sample preparationThe oligo mixture, including a biotin-labelled target-specific DNA oligomer (1:100) and oligos covering other sites (1:30), was annealed to HIV-1 virion RNA at 65 °C for 5 min and then cooled down to room temperature, as described previously42. Samples were digested by nuclease S1 (Invitrogen) for 2 h at room temperature followed by phenol:chloroform purification as previously described42. The biotin-labelled target DNA:RNA duplex was recovered by using Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific) following the manufacturer’s instructions. For RNase T1 digestion, about 200 ng of denatured (95 °C for 2 min and snap cooling at 4 °C) HIV-1 RNA (obtained by modified RNase protection assay) was digested with 50 units of RNase T1 (Worthington) at 37 °C for 2 h and dried in a SpeedVac system (Thermo Fisher Scientific).
LC–MS/MSThe LC–MS/MS analysis was performed using a BEH C18 column (1.7 µm, 0.3 mm × 150 mm, Waters) with Ultimate 3000 ultra high performance liquid chromatography (Thermo Scientific) coupled to the Synapt G2-S (Waters) mass spectrometer as described previously42. The gradient chromatography was performed at 5 µl min−1 flow rate using mobile phase A (8 mM TEA and 200 mM HFIP, pH 7.8 in water) and mobile phase B (8 mM TEA and 200 mM HFIP in 50% methanol) at 60 °C. The gradient consists of an initial hold at 3% B for sample loading, followed by ramping to 55% B in 70 min, 99% in 2 min with 5 min hold before re-equilibration (30 min) at 3% B for initial conditions. The resolved digestion products in the chromatographic eluent were detected in negative ion mode through electrospray ionization on a Synapt G2-S (quadrupole time-of-flight) mass spectrometer operating in sensitivity mode (V-mode). The electrospray ionization conditions included 2.2 kV at capillary, 30 V at sample cone while maintaining source and desolvation temperatures at 120 °C and 400 °C, and gas flow rates at 3 l h−1 and 600 l h−1, respectively. A scan range of 545–2,000 m/z (0.5 s) and 250–2,000 m/z (1 s) was used for first (MS) and second (MS/MS) stage data acquisition. The top three most abundant ions in the first stage were selected for fragmentation for MS/MS using m/z dependent collision energy profile (20–23 V at m/z 545; 51–57 V at m/z 2,000) before excluding them for 60 s using the dynamic exclusion feature.
LC–MS/MS data processingThe m/z values of the RNase T1 digestion products (and their fragment ions) of a 40-base-long HIV-1 RNA sequence were predicted using Mongo Oligo mass calculator. Manual identification and assignment of m6A modification was made by scoring for ~14 Da mass shift of the theoretically expected oligonucleotides (following cleavage at the 3′ end of guanosine) in the modified RNA compared with the unmodified version. A set of controls was used to assign the m6A modification at positions 8,975 and 8,989, respectively.
Site-directed mutagenesisgBlocks (Integrated DNA Technologies) with single or combination mutations of m6A sites were introduced to the HIV-1 vector pNL4-3 for the mutant plasmids. For each mutant plasmid, 500 ng pNL4-3 and 100 ng gBlocks were digested with NcoI-HF and BamHI-HF and ligated for 30 min at room temperature using T4 quick ligation (NEB). The sequences of the mutant plasmids were confirmed by Sanger sequencing.
The mutations were designed based on the following rationales to minimize changes in protein function or RNA structure due to the mutation. The A8079G mutation is situated in the overlapping region of rev and env genes, designed to be a silent mutation for Rev but inducing the substitution of glutamine with glycine at position 771 in Env. We selected this mutation because this amino acid substitution was shown to only moderately reduce HIV-1 fitness in vitro89. A8975C and A8989T are located in the 3′ UTR (U3) of the HIV-1 RNAs. These mutations were chosen to preserve the RNA structures predicted by a minimal free energy structure prediction tool90. Although the structures used to design these mutations remain to be validated, A8079G, A8975C and A8989T mutant viruses replicate normally, exhibiting insignificant or only marginal differences in various features of their RNAs—including m6A methylation, alternative splicing, 3′ poly(A) tail, and translation of viral RNAs—suggesting an insubstantial impact of the mutations themselves.
Digital quantitative PCR for total HIV-1 RNAViral RNA production was measured by RT–PCR and DRS analysis. An equal amount of total cellular RNA for each sample was used for RT and complementary DNA generation. The QuantStudio 3D Digital PCR System (Applied Biosystems) was used with appropriate consumables provided by the manufacturer, including the QuantStudio 3D Digital PCR Master Mix v.2 and TaqMan 5′-6 FAM or VIC probe (Applied Biosystems). The primers used are listed in Supplementary Table 9.
Western blot analysisCollected cells were lysed in RIPA 1× buffer (Abcam) with a protease inhibitor cocktail (Sigma-Aldrich) and incubated for 30 min on ice. The cell lysates were centrifuged at 12,000g for 15 min at 4 °C and the supernatant was transferred to a fresh tube and mixed with an equal volume of Laemmli 2× buffer (Bio-Rad). The mixture was then incubated at 95 °C for 10 min. The proteins were separated on sodium dodecyl sulfate–polyacrylamide gels and then transferred to a nitrocellulose membrane (Bio-Rad). The membranes were washed in 1× PBS, Tween 20 (PBST) (10 mM sodium phosphate, 0.15 M NaCl and 0.05% Tween 20) and blocked in 5% milk in 1× PBST for 1 h. Primary and secondary antibodies were diluted at 1:1,000 and 1:5,000, respectively, each in 5% milk in 1× PBST. The signals were visualized by chemiluminescence. The primary antibodies used were HIV-1 p24 (NIH AIDS Reagent Program, cat. no. ARP-6458,1:1,000), gp41 (NIH AIDS Reagent Program, cat. no. ARP-11391,1:500), Vif (NIH AIDS Reagent Program, cat. no. ARP-6459,1:500), GAPDH (Abcam, cat. no. ab8245,1:1,000) and anti-mouse (Promega, cat. no. W4021,1:5,000) for the secondary antibody.
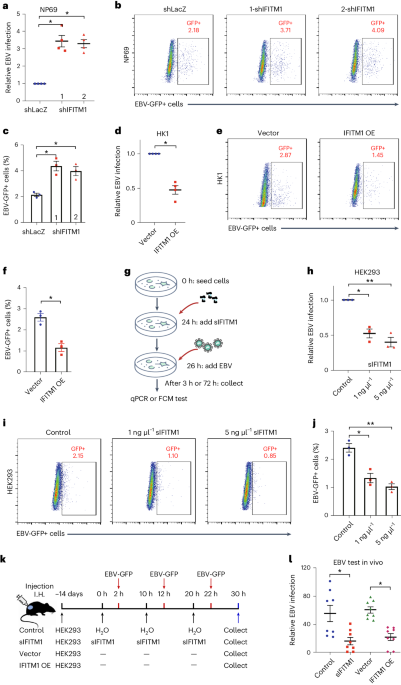
Measuring HIV-1 infectivity using GFP reporter cellsAn equal amount of virus stock (pg) was used to infect GHOST R3/X4/R5 cells (ARP-3943, NIH HIV reagent program) in 6-well plates91. After 48 h post-infection, the cells were washed with PBS and fixed. The green fluorescent protein (GFP) expressions for all samples were acquired by the Attune NxT flow cytometer (Thermo Fisher Scientific) and analysed by the FlowJo software (BD Biosciences).
Single cycle infection of CEM-SS cellsA total of 2 × 106 CEM-SS cells (ARP-776, NIH HIV reagent program) were infected with WT or triple mutant virus at 2 multiplicity of infection (MOI) in 1 ml Roswell Park Memorial Institute (RPMI) 1640 with 1% penicillin/streptomycin (P/S) and 10% foetal bovine serum (FBS). Cells were incubated with viruses for 1 h, swirling every 20 min, and then transferred to T25 flask with 10 ml RPMI 1640 (1% P/S and 10% FBS). At 24 h post-infection, the cells were washed with PBS and the culture medium was exchanged with RPMI 1640 medium with drugs (1% P/S, 10% FBS, 100 nM T20 and 100 nM IDV). At 96 h post-infection, the single cycle infected cells were collected and total cellular RNAs were extracted with TRI Reagent (Sigma-Aldrich, T9424), following the manufacturer’s instructions.
Jurkat cell infectionA total of 6 × 106 Jurkat cells (ARP-177, NIH HIV reagent program) were infected with WT or triple mutant virus at 1 MOI (first experiment) or 2 MOI (second and third experiments) in 2 ml RPMI 1640 (1% P/S and 10% FBS) for 1 h, swirling every 20 min. Cells were then transferred to T75 flask, adding RPMI 1640 (1% P/S and 10% FBS) up to 30 ml. At 24 h post-infection, the cells were washed with PBS and the culture medium was exchanged with fresh RPMI 1640 (1% P/S and 10% FBS). At 96 h post-infection, total cellular RNAs were extracted with TRI Reagent (Sigma-Aldrich, T9424), following the manufacturer’s instructions.
Machine learning: determining m6A modifications per-read per-position basisThe goal is to build machine learning models that determine whether an HIV RNA molecule has m6A modifications on a per-read and per-position basis. The classification is based on the read-level P value output from Tombo-MSC. The source code is available at ref. 92.
Read-level P value patterns analysisTombo-MSC uses IVT data to adjust the expected current signal levels and generates per-read, per-position P values for current signal difference of target RNA (native HIV-1 RNA) reads. We found that Tombo-MSC’s per-read P values were highly reproducible when a sufficient number of IVT canonical control reads were used (for example, r2 > 0.999 with >20,000 IVT reads; Extended Data Fig. 2c) and when tested in our repeated experiments using four sets of virion RNAs that were prepared independently of each other (Extended Data Fig. 2d). We also found a consistent pattern of per-read P value distribution near the three m6A sites (positions 8,079, 8,975 and 8,989) on native HIV-1 RNA, showing a shift of the d values (or median P value) to upstream of the m6A sites at positions N−4 to N1 (N0, m6A site) (Fig. 5a(ii) and Extended Data Fig. 9e). The patterns were consistent between native HIV-1 RNAs and m6A-control RNAs. Here we developed new read-level binary classification methods m6A based on the read-level P values at positions N−4 to N1 (m6Arp models) for the three predominant m6A sites. Additional positions (including N2 to N5) contributed only negligibly to the model accuracy.
Preparing control RNAsWe built three separate models to detect modifications at positions 8,079, 8,975 and 8,989. We generated approximately 1 kb long positive control RNAs using three synthetic RNA oligos, each harbouring m6A at 8,978, 8,975 and 8,989, respectively (Supplementary Table 2 and Extended Data Fig. 9a–d). The 8,079, 8,975 and 8,989 models used DRS reads of these RNAs (8978m6A+, 8975m6A+ and 8989m6A+ datasets) as positive-labelled training data. In parallel, IVT RNA reads that cover the same 1 kb region were used as negative-labelled training data. We also tested full-length IVT RNA and F2 IVT RNA reads as negative-labelled training data. These negative-labelled training data showed only negligible difference in the performance of the m6Arp models.
Synthetic RNA oligos were custom synthesized by Horizon Discovery (Extended Data Fig. 9a). Positive-control RNAs were generated by ligating carrier IVT RNA to synthetic RNA oligos (Extended Data Fig. 9b); the ligated RNA aligns to approximately 1 kb of the 3′ end of HIV-1 RNA. The 3′ end of carrier IVT RNA and the 5′ end of phosphorylated synthetic RNA were joined by ligating these two RNAs. The ligated RNA was then subjected to poly (A) tailing using Escherichia coli poly(A) polymerase (NEB). The carrier IVT RNA were generated as described in ‘Preparation of IVT RNA controls’ section above using DNA templates generated by PCR using primers shown in Supplementary Table 3.
Selecting Fisher optionsModels trained with Tombo-MSC P values generated with Fisher 0, 1, 2 and 3 showed varying levels of the AUROC, FPR and FNR (Extended Data Fig. 9f). Of these, the ‘Fisher = 0’ option showed the best AUROC values for all three models (models 8079, 8975 and 8989) ranging from 0.95 to 0.97, with 8.40–10.80% FPR and 8.90–12.40% FNR (Supplementary Table 7).
Model selectionAll our machine learning models were linear support-vector classifiers, implemented using the scikit-learn Python package (v.0.23.2) (ref. 93). Models used default scikit-learn settings, except where noted. We selected support-vector classifiers in consideration of their potentially high generalizability and resistance to over-fitting due to their small number of parameters (n + 1, where n is the number of features). The magnitude of a coefficient in the model is a measure of the relevance of the corresponding feature to the classification problem. Coefficients of our trained models are reported in Supplementary Table 7. The positive and negative classes were weighted to achieve a balanced comparison. Other than this weighting, models used default parameters as listed on the scikit-learn documentation page. As an alternative to the support-vector classifier described here, an unsupervised learning model based on the DBSCAN algorithm was also assessed. It demonstrated a similar FPR and FNR to the support-vector classifier (data not shown).
Five-fold cross-validation for the accuracy of the modelThe model accuracy was first assessed using fivefold cross-validation94. In brief, to assess the accuracy of a model with dataset T, the dataset is partitioned at random into five equal-size subsets, T1, T2, T3, T4 and T5. Five new models are trained, each on its own four-fifths of the data. For example, Model M1 is trained using the combined data of T2, T3, T4 and T5, then its FNR and FPR are computed using the reserved partition T1. The FNRs and FPRs from all five folds are averaged to estimate the model’s performance on future unseen data.
Linearity of quantification analysisTo evaluate the ability of each model to enumerate m6A positive and negative reads at the test site represented in their training data, we tested the linearity of quantification of the three models. For the three testing sites (positions 8,079, 8,975 and 8,989), we generated five sets of mixed data each having positive to negative reads ratios of 0:100, 25:75, 50:50, 75:25 and 100:0 (Extended Data Fig. 9g) and assessed the ability of the three new models to quantitate m6A modifications. The quantitation results were directly proportional to the fraction of m6A positive control reads in premixed controls with expected FPR and FNR at both ends of mix ratios (Extended Data Fig. 9g). After adjusting for each model’s FPR and FNR, the output showed nearly complete matches to the expected values.
Nucleotide sequence conservation near the major m6A sitesThe HIV-1 B subtype sequences corresponding to A8079, A8110, A8975 and A8989 of the NL4-3 strain (RNA) were extracted from the HIV sequence database (https://www.hiv.lanl.gov/). The sequence logo plots were generated using the ggseqlogo R package (v.0.1).
HIV-1 RNA splicing analysisReads were aligned using minimap2 (v.2.24) against the HIV genome AF324493.2 from the NCBI in spliced mapping mode using a k-mer size of 14. Unmapped reads, alignments with a quality lower than 30, and alignments were discarded using SAMtools (v.1.6). Reads were screened for potential splicing donor (SD) and splicing acceptor (SA) sites by identifying exon start/end positions from BED files. SD and SA sites46 are shown in Figs. 1c,4a and 6a. Reads presenting the same combination of splice junction and exotic sequences were grouped together and counted. If the potential splicing sites do not match to the SD/SA sites46, the reads were annotated as unclassified. Annotation was performed according to the convention established in ref. 46 or for potential spliced isoforms, to the open reading frame encountered in the read. The relative levels of spliced viral isoforms were quantified using absolute counts compared with the WT. Splicing donor and splicing acceptor usage were calculated using absolute counts after screening of known SD/SA sites from BED files. The reads were classified as spliced when their splice junction include either D1 or D1c splicing donor site. If they have an A7 splicing acceptor site, they were further classified into CS. Then, the rest were classified into PS. The poly(A) tail length of each classified group was estimated using the Nanopolish (v.0.14.0) polya module with default parameters.
IVT read depth analysisSix sets of IVT data were generated by randomly choosing 500, 1,000, 5,000, 10,000, 20,000, 30,000 and 40,000 reads from a pool of a total 50,000 half-length IVT reads. Each of these IVT sets was tested as a canonical control for Tombo analysis (Fishers 0). A total of 384 reads of HIV-1 virion RNA were used as a sample dataset. The averaged P values for each position were tested with non-parametric pairwise comparison using the ggstatsplot (v.0.9.1) R package. A P value <0.05 was considered significant.
Statistical analysisStatistical analysis was performed using GraphPad Prism9 (v.9.5.0) or R package (v.4.0.2), and detailed statistical tests used are indicated. All averaged data include error bars that denote s.d., with single data points shown. A P value <0.05 was considered significant. We provide triplicated (or quadruplicated) experimental data of biologically independent samples. No statistical methods were used to predetermine sample sizes, but the sample size of n = 3 or n = 4 routinely provides sufficient statistical power (when present) in our study utilizing accurate and highly reproducible assays and molecular biology experiments. These number of samples are commonly used in molecular biology publications to provide statistical conclusions, as well as to address the rigour and reproducibility. Data collection and analysis were not performed blind to the conditions of the experiments. Experimental groups were determined on the basis of the experimental hypothesis (for example, the impact of mutations or RNA isoforms) and all the experimental data and sequencing data that pass the data exclusion criteria were used without any additional selection. Data collection and analysis were not performed blind to the conditions of the experiments.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
留言 (0)