The research conducted in this study complies with all relevant ethical regulations. Experiments using genetically modified mice and recombinant DNA were approved by the Animal Care and Use Committee and the Recombinant DNA Experiment Committee of Nagoya University (approval nos. R230006, 166-101 and 169-113).
Plasmid and siRNA
The full-length human TFIIS cDNA (clone ID 6470030, Horizon Discovery) was cloned into pEGFP-C1 (Clontech) containing the N-terminal Strep-V5-tag. The TFIIS cDNAs were rendered insensitive to TFIIS siRNA by introducing the underlined silent mutations (5′- aacagGaaAgaCgaAacaaat-3′). Cloning and site-directed mutagenesis were performed using KOD-Plus-Neo (TOYOBO). All constructs were verified by Sanger sequencing. Plasmid DNA and siRNA transfections were performed using X-tremeGENE HP (Roche) and Lipofectamine RNAiMAX (Thermo Fisher Scientific), respectively, according to the manufacturer’s instructions. The siRNA target sequences used in this study are listed in Supplementary Table 7.
Cell lines and culture
HeLa cells (laboratory stock) and 293FT cells (Thermo Fisher Scientific) were maintained in Dulbecco’s modified Eagle medium (DMEM, FUJIFILM) supplemented with 10% fetal bovine serum (FBS, Invitrogen) and antibiotics, unless otherwise noted. Mycoplasma testing was performed routinely. Cell lines stably expressing Strep-V5-GFP-TFIIS were obtained by selecting HeLa cells transfected with pEGFP-C1-Strep-V5-GFP-TFIIS plasmid in medium containing 500 μg ml−1 G418 (Nacalai Tesque). HeLa ΔCSB cells were transfected with pEGFP-C1-Strep-V5-GFP-CSB plasmid in medium containing 500 μg ml−1 G418 (Nacalai Tesque). Unless otherwise indicated, the following doses of chemicals were used: CB5083 (10 μM, Selleck), DRB (100 μM, TCI), epoxomicin (1 μM, FUJIFILM), formaldehyde (600 μM, Nacalai Tesque), MG262 (1 μM, AdipoGen Life Sciences), NMS873 (10 μM, Sigma-Aldrich) and triptolide (3 μM, AdipoGen Life Sciences).
Genome editing using CRISPR-Cas9 technology
ΔCSB, ΔCSA, ΔUVSSA and POLR2AKR cells have been described previously31. To edit the FANCA gene, HiFi Cas9 nuclease V3 (Integrated DNA Technologies) was mixed with CRISPR RNA (crRNA):trans-activating CRISPR RNA (tracrRNA) complex. The mixture was electroporated into HeLa cells using 4D-Nucleofector (Lonza). Cells were recovered by DMEM with 10% FBS and cultured on a 35-mm dish for 24 h. Single-cell clones were isolated using limiting dilution in 96-well plates. To edit the XPA gene, a guide RNA (gRNA) coding sequence was cloned into the PX459 vector55. HeLa cells were transfected with the plasmid using Lipofectamine 2000 (Thermo Fisher Scientific). Cells were selected for 48 h in medium containing 2 μg ml−1 puromycin (Nacalai Tesque). Single clones were isolated by limiting dilution. Mutations of gene-edited cells were confirmed by Sanger sequencing. The crRNA, gRNA, and primer sequence information is listed in Supplementary Table 7.
Immunochemical methods
To analyse whole cell lysate, cells were dissolved in Laemmli sodium dodecyl sulfate (SDS) sample buffer (50 mM Tris pH 6.8, 10% glycerol, 100 mM dithiothreitol (DTT), 2% SDS, 0.1% bromophenol blue) and cleared by centrifugation. Immunoprecipitation with anti-RPB1-phospho-Ser2-CTD antibodies was performed as described in ref. 31. Briefly, cells were suspended in EBC buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.5% NP-40, 2 mM MgCl2) supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Nacalai Tesque). After centrifugation for 5 min at 4 °C, the supernatants were collected (nuclear/cytoplasmic fraction), and the pellets were further incubated in EBC buffer with Benzonase (Merck) for 1 h at 4 °C, followed by centrifugation (chromatin fraction). The extract was then incubated with anti-RPB1-phospho-Ser2-CTD antibodies (ab5095, abcam) and protein A agarose beads (Milipore, #16-157) for 3 h on a rotator at 4 °C. After an extensive wash in lysis buffer, the immunoprecipitates were eluted with 2× Laemmli sample buffer. A Strep-Tactin pulldown assay was carried out as described in ref. 56. Briefly, 293FT cells were transfected with plasmid harbouring Strep-Myc-tagged ubiquitin using X-tremeGENE HP (Roche) according to the manufacturer’s instructions. Cells were lysed in denaturing buffer (20 mM Tris, pH 7.5, 50 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.5% NP-40, 0.5% sodium deoxycholate and 0.5% SDS) supplemented with protease and phosphatase inhibitors. After sonication and clearing of the lysates, the supernatant was incubated with Strep-Tactin Sepharose (IBA Lifesciences) for 3 h at 4 °C. After extensive washing with denaturing buffer, the precipitates were eluted with 2× Laemmli sample buffer. Proteins were resolved by SDS–polyacrylamide gel electrophoresis (PAGE). Resolved protein samples were transferred to polyvinylidene fluoride membrane for immunodetection. The antibodies used for immunochemical experiments are listed in Supplementary Table 8.
MS analysis
Enriched proteins using anti-RPB1-phospho-Ser2-CTD antibodies (ab5095, abcam) or anti-TFIIS antibodies (302–239A, Bethyl Laboratories) were digested after alkylation using the filter-aided sample preparation protocol57. Briefly, the immunoprecipitates were dissolved in SDS buffer (0.1 M Tris pH 8.5, 2% SDS, 100 mM DTT) and boiled for 5 min. The proteins were diluted in 8 M urea in 0.1 M Tris pH 8.5, and loaded onto 30-kDa Vivacon 500 centrifugal units (Sartorius). The buffer was replaced by washing with urea buffer, and the proteins were alkylated with 50 mM iodoacetamide for 20 min at 25 °C in the dark. The proteins were then digested with Lys-C (FUJIFILM) and trypsin (Thermo Fisher Scientific) overnight at 37 °C. The resulting peptides were purified using C18 tips (AMR). The peptides from the immunoprecipitates were analysed by liquid chromatography mass spectrometry (LC-MS) using an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) coupled to an UltiMate3000 RSLCnano LC system (Dionex Co.) using a nano-HPLC capillary column (150 mm × 75 m i.d., Nikkyo Technos Co.) via a nanoelectrospray ion source. Reversed-phase chromatography was performed with a linear gradient (0 min, 5% B; 75 min, 35% B) of solvent A (2% acetonitrile with 0.1% formic acid) and solvent B (95% acetonitrile with 0.1% formic acid) at an estimated flow rate of 300 nl min−1. A precursor ion scan was carried out using a 400–1,600 mass-to-charge ratio (m/z) before MS/MS analysis. Tandem MS was performed by isolation at 0.8 Th with the quadrupole, higher-energy collisional dissociation (HCD) fragmentation with a normalized collision energy of 30%, and rapid scan MS analysis in the ion trap. Only those precursors with charge state 2–6 were sampled for MS2. The dynamic exclusion duration was set to 15 s with a 10-ppm tolerance. The instrument was run in top speed mode with 3-s cycles. All experiments were performed in the data-dependent acquisition mode to automatically isolate and fragment top ten multiply-charged precursors (+2, +3, +4 and +5) according to their intensities. Former target ions were dynamically excluded for 15 s, and all experiments were acquired using the positive polarity mode. The full scan resolution was set to 70,000, and the mass range was set to m/z 350–1,400. The full scan ion target value was set to 3 × 106, allowing a maximum fill time of 60 ms. HCD fragment scans were acquired with the optimal setting for parallel acquisition using a 1.6 m/z isolation window and a normalized collision energy of 27.
Proteomic data analysis
Raw data were analysed with MaxQuant software (version 1.6.0.1). A UniProt database for the human proteome (Proteome ID UP000005640) was used to search for precursor ions and MS/MS spectra using the Andromeda search engine58. Carbamidomethylation of cysteines was searched as a fixed modification, and oxidation of methionines and acetylation of protein N termini as variable modifications. Enzyme specificity was set to trypsin and Lys-C, and a maximum of two missed cleavages were allowed for searching with a precursor mass tolerance of 4.5 ppm and a fragment mass tolerance of 20 ppm. A false discovery rate (FDR) of 0.01 for proteins and peptides and a minimum peptide length of seven amino acids were required. Quantification in MaxQuant was performed using the built-in label-free quantification (LFQ) algorithm. LFQ intensities were analysed with the statistical software package Perseus (version 1.6.0.7). The logarithmized LFQ intensities of the compared conditions were filtered to have two valid values in at least one sample group. Missing values were imputed by creating a normal distribution with a width of 0.3 relative to the standard deviation of the measured values and a 1.8-standard-deviation downshift of the mean. Significant differences in protein abundance were assessed by an unpaired two-sided t-test with permutation-based FDR < 0.05. The proteomic data were visualized using R (version 4.0.3) in RStudio (version 2022.02.3 Build 492).
Measurement of BrU incorporation by flow cytometry
For the measurement of RRS after formaldehyde DNA damage, cells were treated with 1,200 μM HCHO for 1 h, followed by 12-h incubation for recovery. Cells were labelled with 1 mM 5-bromo-uridine (BrU) for 1 h, followed by fixing in 4% formaldehyde for 15 min, then permeabilized with PBS containing 0.25% Triton X-100 for 10 min on ice. After washing with PBS containing 0.05% Tween20, the cells were stained with 2 μg ml−1 Alexa Fluor 647 NHS Ester (A37573, Thermo Fisher Scientific) for 30 min at room temperature. After washing with PBS containing 0.05% Tween20, stained cells were mixed at a 1:1 ratio with unstained cells. These cells were then stained with Alexa Fluor 488 anti-BrdU antibodies, and the nuclei were stained with 1 μg ml−1 4′,6-diamidino-2-phenylindole (DAPI; D523, DOJINDO). Data were acquired on a CytoFLEX S FACS analyser (Beckman Coulter) by CytExpert (version 2.0) and analysed with FlowJo software (version 10.8.1, BD). The gating strategy is provided in Extended Data Fig. 10.
RRS assay using 5-ethynyluridine
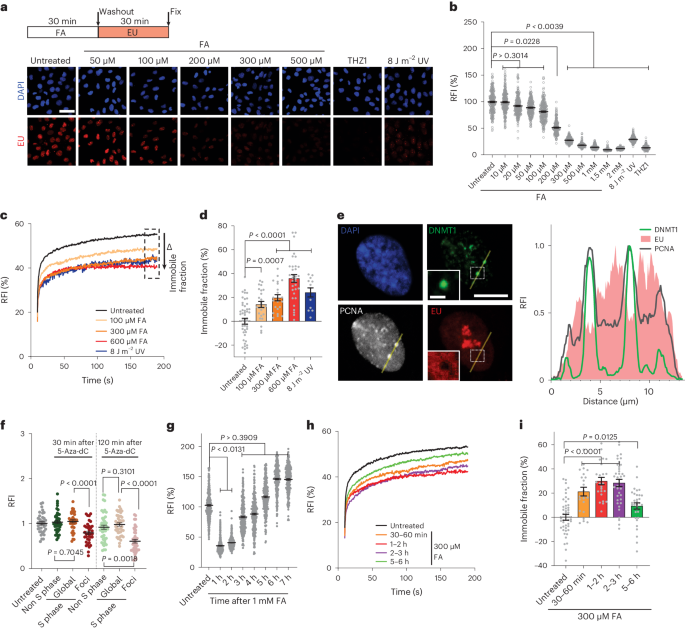
Details of the assay have been described previously31. Briefly, cells were cultured in DMEM supplemented with 10% FBS and plated in plastic 96-well plates. Cells were treated with formaldehyde or irradiated with UV light (254 nm UVC), followed by incubation for RRS. The RRS levels were measured by ethynyluridine (EU) incorporation. Recovered cells as well as untreated cells were incubated for 2 h in medium supplemented with 100 μM 5-ethynyluridine (5-EU, Thermo Fisher Scientific), followed by fluorescent-azide conjugation (Click-chemistry, Thermo Fisher Scientific). The cells were fixed and permeabilized for 20 min in PBS containing 2% formaldehyde and 0.5% Triton X-100. After washing with PBS, the cells were then incubated with coupling buffer with 10 μM Alexa Fluor 488 azide (A10266, Thermo Fisher Scientific), 50 mM Tris-HCl (pH 7.3), 4 mM CuSO4, 10 mM sodium ascorbate and 30 ng ml−1 DAPI (D523, DOJINDO) for 60 min, followed by washing with PBS containing 0.05% Tween20. Nuclear fluorescent image acquisition and data processing were automated with HCS Studio 2.0 software in the ArrayScan VTI system (Thermo Fisher Scientific).
Clonogenic survival assays
Cells were seeded in six-well plates and treated with formaldehyde for 2 h or irradiated with UV light. After removing the formaldehyde, cells were allowed to grow for nine days. Cells were fixed with methanol and stained with crystal violet, and colonies with more than 50 cells were counted.
Animals
C57BL/6JJcl mice were purchased from CLEA Japan. The animals were kept under conditions of 23 ± 1 °C, 50 ± 5% humidity and a 12-h:12-h light:dark cycle. They were fed a standard pellet diet (CE-2, CLEA Japan) and tap water ad libitum. Both male and female mice were used. In Fig. 6b,d–f and Extended Data Figs. 8a,b and 9a–c, mice at three to four weeks of age were analysed. In Fig. 6c, mice at two weeks to one year of age were analysed. In Extended Data Fig. 7a, mice at two weeks to three years of age were analysed. In Extended Data Fig. 7b–d, mice at two weeks of age were analysed. In Extended Data Fig. 7e, mice at two weeks to seven months of age were analysed.
Genome editing of mouse embryos
Adh5−/− and Aldh2+/KI mice have been described previously in ref. 11. To generate Csb-deficient mice, the following reagents were purchased: Cas9 Nuclease V3, tracrRNA and crRNA (Integrated DNA Technologies). Pronuclear-stage mouse embryos were prepared by thawing frozen embryos (CLEA Japan), then culturing them in potassium simplex optimized medium (KSOM, ARK Resource). For electroporation, 100–150 embryos (1 h after thawing) were placed into a chamber with 40 μl of serum-free medium (Opti-MEM, Thermo Fisher Scientific) containing 100 ng μl−1 Cas9 Nuclease V3 and 100 ng μl−1Csb gRNA. They were then electroporated with a 5-mm gap electrode (CUY505P5, NepaGene) in a NEPA21 Super Electroporator (NepaGene). The pulses for the electroporation had a voltage of 225 V, pulse width of 1 ms for mouse embryos, pulse interval of 50 ms, and the numbers of pulses was 4. The first and second transfer pulses had a voltage of 20 V, pulse width of 50 ms, pulse interval of 50 ms, and the number of pulses was 5. Mouse embryos that developed to the two-cell stage after electroporation were transferred into the oviducts of female surrogates anaesthetized with sevoflurane or isoflurane (Mylan). gRNA sequence information is listed in Supplementary Table 7.
Mice haematological analysis
Red blood cells (RBCs), haemoglobin concentration and haematocrit were measured using an IDEXX ProCyte Dx system (IDEXX Laboratories).
Bone marrow cell isolation and FACS analysis
Bone marrow cells were flushed from mice femurs and tibias using a 26-G needle followed by passing through a cell strainer in Ca2+- and Mg2+-free Hank’s buffered salt solution (HBSS, Gibco) supplemented with 1% heat-inactivated bovine serum (Gibco). The RBCs were lysed by resuspending the cells in RBC lysis buffer (eBioscience) for 5 min on ice. The cells were filtered through a 70-μm cell strainer to obtain a single-cell suspension. The number of cells was measured with a haemocytometer. Lineage-depleted mouse bone marrow cells were prepared using a Direct Lineage Cell Depletion Kit (Miltenyi Biotec). The antibodies used for FACS analysis were as follows: FITC-conjugated lineage cocktail, CD41, FcεRIα, CD117, Sca-1, CD48, CD150, CD135, CD127, CD16/32 and CD34. Antibody staining was performed at 4 °C for 20 min. Dead cells were excluded by staining with 7-AAD (BioLegend). Data were acquired on a CytoFLEX S FACS analyser (Beckman Coulter) by CytExpert 2.0 and analysed with FlowJo v10.8.1. Antibodies used for FACS analysis are listed in Supplementary Table 8.
DPC quantification and DPC-seq
Protein-conjugated DNA was isolated using a conventional KCl/SDS precipitation method32. The cells were lysed in SDS buffer (2% SDS; 20 mM Tris, pH 7.5) followed by sonication using a Covaris M220 system (5% duty factor, 200 cycles per burst, 16 min at 25 °C in microTUBE). The sonicated samples were incubated with KCl buffer (200 mM KCl, 20 mM Tris pH 7.5) on ice for 5 min. After centrifugation at 20,000g for 5 min at 4 °C, the supernatant was collected as soluble DNA. Pellets were washed five times by adding KCl buffer followed by incubation at 55 °C for 5 min. After centrifugation, the pellets were dissolved in KCl buffer with 0.04 mg ml−1 Proteinase K and incubated at 55 °C for 3 h. After centrifugation at 20,000g for 5 min at 4 °C, the supernatant was purified using a MinElute PCR purification kit (QIAGEN). DNA concentration was measured using Qubit dsDNA HS. The amount of DPCs was calculated as the ratio of precipitated DNA to total DNA (precipitated plus soluble DNA).
In the experiments shown in Extended Data Fig. 4d,e, the cells were synchronized as described in ref. 59. Cells were grown in the presence of 2 mM thymidine (Sigma-Aldrich) for 24 h, washed with PBS and grown in fresh medium for 9 h. The cells were then cultured in medium with thymidine for 24 h, washed with PBS, and grown in medium with 0.5 μg ml−1 nocodazole (Sigma-Aldrich) for 11 h. After release from the nocodazole block, the cells were treated with formaldehyde for 1 h, washed with PBS, and recovered for 4 h in the presence of thymidine to block S-phase entry. Cell synchronization was confirmed by incorporation of 5-ethynyl-2′-deoxyuridine (EdU, Thermo Fisher Scientific). Briefly, 2 h after release from the nocodazole block, cells were labelled with 5 μM EdU for 5 h in the presence of thymidine, followed by fixing in 4% formaldehyde for 10 min and permeabilization with PBS containing 0.5% Triton X-100 for 5 min. After washing with PBS, the cells were then incubated with coupling buffer with 10 μM Alexa Fluor 488 azide (A10266, Thermo Fisher Scientific), 50 mM Tris-HCl (pH 7.3), 4 mM CuSO4 and 10 mM sodium ascorbate. The cells were imaged on a BZ-X800 microscope (Keyence).
For the detection of histone-DPCs, cells were incubated in EBC-2 buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.5% NP-40, 5 mM EDTA, 0.5 mM EGTA) supplemented with protease inhibitor cocktail (Roche) for 20 min on ice. After centrifugation at 10,000g for 5 min at 4 °C, the pellets were dissolved in incubation buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 5 mM EDTA, 0.5 mM EGTA, 0.1% BSA) supplemented with protease inhibitor cocktail (Roche), and sonicated using a Covaris M220 system (5% duty factor, 200 cycles per burst, 16 min at 7 °C in microTUBE). After centrifugation at 20,000g for 10 min at 4 °C, the supernatant was incubated with anti-H2B (ab1790, abcam) antibodies and protein A and G magnetic beads (Bio-Rad) for 12–16 h on a rotator at 4 °C. After extensive washing, the beads were incubated with SDS buffer (2% SDS, 20 mM Tris pH 7.5) for 15 min at 30 °C. The eluted supernatant was subjected to DPC isolation.
Purified DNA was subjected to NGS. NGS libraries were generated using NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB) or MGIEasy Universal DNA Library Prep Set (MGI). The prepared libraries were sequenced on a HiSeqX platform (Illumina) with 150-bp paired-end reads, a HiSeq2000 platform (Illumina) with 80-bp single-end reads, a DNBSEQ-G400 platform (MGI Tech) with 150-bp paired-end reads or a DNBSEQ-T7 platform with 150-bp paired-end reads.
The method is available from the Protocol Exchange web site60.
Sequence data analysis
NGS sequence reads were adaptor-trimmed using fastp61 (version 0.21.0) and aligned to the GRCh38 (human) or mm10 (mouse) reference genomes using bowtie2 (ref. 62) (version 2.3.4.3). Uniquely mapped reads were sorted using samtools63 (version 1.15.1), and duplicates were removed using picard MarkDuplicates (version 2.25.0, Broad Institute). The sequence data are listed in Supplementary Table 9.
Mapped reads were visualized using Integrated Genome Viewer64 (IGV; version 2.14.1). Read coverage and scores were calculated and visualized using deepTools65 (version 3.5.1). The mapped reads on a ribosomal RNA unit were visualized according to ref. 66. Spearman’s rank-correlation coefficients between experiments were calculated using deepTools.
To analyse the relationship between transcription level/gene length and DPC repair kinetics, the longest transcript for each gene was extracted from the corresponding reference genomes, and genes with regions overlapping another gene were excluded using bedtools67 (version 2.27.1). The gene expression profile of HeLa cells was obtained from the Expression Atlas of EMBL-EBI (https://www.ebi.ac.uk/gxa/experiments/E-MTAB-2706/Results) and the gene expression profile of lineage-depleted cells from mice bone marrow was acquired from RNA-seq data in this study (Supplementary Table 10). For read quantification, mapped reads to reference genomes were counted using HTseq68 (version 2.0.2) or bedtools. After excluding mitochondrially encoded genes (in both human and mouse) as well as those on sex chromosomes (in mouse), the read counts were normalized according to their gene length, and the sum was rescaled to 1 × 106 per sample.
RNA-seq
Total RNA from lineage-depleted mouse bone marrow cells (n = 3) was extracted using an RNeasy Kit (QIAGEN). RNA was quantified with a Qubit RNA BR Assay Kit (Life Technology), and integrity was evaluated with Agilent Bioanalyzer 2100 (Agilent Technologies). RNA-seq libraries were constructed with an MGIEasy RNA Directional Library Prep Set (MGI) according to the manufacturer’s instructions. Libraries were sequenced with 150-bp paired-end reads on a DNBSEQ-G400 sequencer (MGI Tech). Low-quality reads and adapter sequences were trimmed using fastp software61. The reads were mapped to the GRCm38 reference genome using HISAT269 (version 2.1.0), followed by transcript assembly and quantification using StringTie70 (version 1.3.4d).
Statistics and reproducibility
The statistical tests used are indicated in the figure legends. No statistical method was used to predetermine sample size. The sample sizes chosen are consistent with our previous publications. Data were excluded from analysis only in cases of obvious technical failure. Python (version 3.9.13), R (version 4.0.3), JMP (version 16.1.0) and Excel (version 16.80) were used to generate graphs and perform statistical analyses. Most experiments were replicated. All replication attempts were successful. The numbers of replicate experiments are given in the figure legends or in the figures. Samples were not randomized for this study. This study was not blinded except for colony counting.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this Article.





留言 (0)