
記住我

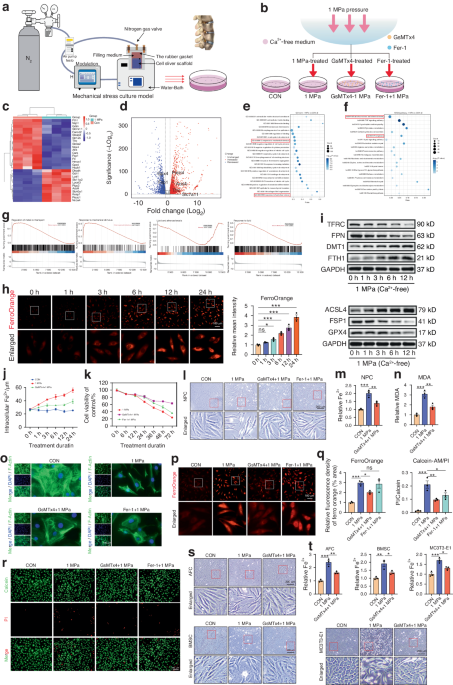
The patients were diagnosed with either ossification of the posterior longitudinal ligament (OPLL) or non-OPLL conditions such as cervical spondylotic myelopathy or radiculopathy without ossification. The diagnosis was based on symptoms, radiological findings from CT, MRI, and X-ray, as well as intraoperative exploration. A standard Anterior Cervical Discectomy and Fusion (ACDF) technique was applied during the surgery. The tissue samples were excised with a Kerrison rongeur, washed twice with saline by a scrub nurse, and transferred to our researcher immediately in wet sterile dressings. After resection, the samples were immersed in sterile saline and transported at 4 °C. The ligaments were rinsed in phosphate buffer saline (PBS) twice and cut into small pieces measuring about 1 mm × 1 mm. These small pieces were evenly spread on the surface of a 6 cm dish and left for 10 min to dry before they were firmly attached to the bottom of the dish. Subsequently, a medium (Mesenchymal Stem Cell Basal Medium, Dakewe, Cat# 6114021) supplemented with 5% serum substitute (EliteGro™-Adv, EliteCell, Cat# EPA-500) and 1% penicillin/streptomycin was carefully placed on the dish to avoid washing away the tissue. The dish was then incubated at 37 °C under 5% CO2. The ligament cells were passaged using trypsin and digested for 1 min when they reached 70%–80% confluency. Only cells at passages 2–5 were used for subsequent experiments. To mimic the hypoxic environment in ligaments, an additional 100 μmol/L of cobalt chloride was added to the culture medium of ligament cells during plasmid transfection and tube formation assays. Fasting morning serum samples were obtained after admission using procoagulant tubes, and the tubes were centrifuged at 3 000 r/min for 5 min to separate the serum. The serum samples were aliquoted and stored at −80 °C. Overall, we collected five OPLL patient tissue samples and five non-OPLL patient samples for cell culture and subsequent in vitro experiments; five ossified and three non-ossified patients’ samples were used for staining (Table S7). and 30 OPLL and 14 non-OPLL patients’ serum samples were used for VEGFA Elisa assay(Table S6). The study was approved by the 905th Hospital of PLA Navy Ethics Committee.
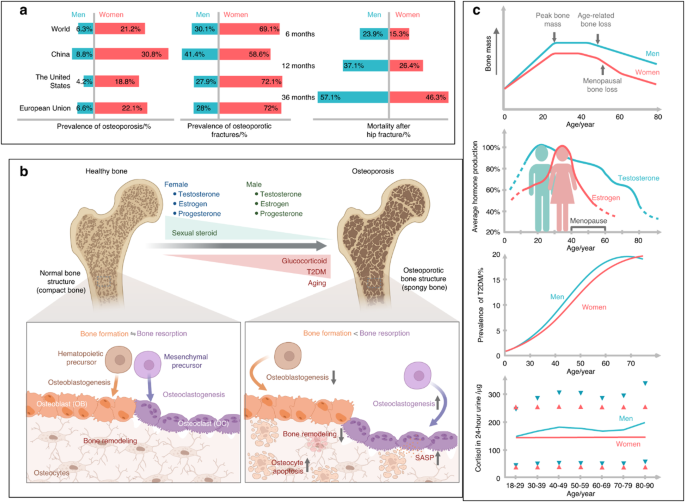
Quantification of ossification and disease severityPlain X-ray (DRX-Nova, Carestream Health), 1.5 Tesla magnetic resonance (MAGNETOM Aera, Siemens Healthineers), and 64-detector row CT (Sensation 64, Siemens) images were obtained from the 905th Hospital of PLA Navy. To quantify the ossification of patients and mice, we used the ossification index (OS index) and the Occupancy Ratio of the spinal canal. In short, the OS index was used to determine the total segments of disks and vertebral bodies involved in ossification. The Occupancy Ratio of the spinal canal was used to assess the maximal ossification thickness to the diameter of the spinal canal on the sagittal section. This study used a modified Japanese Orthopaedic Association (mJOA) scale to evaluate the disease severity of OPLL patients from 3 dimensions: motor dysfunction, sensory impairment, and bladder dysfunction symptoms. The neck disability index (NDI) was used to evaluate disease severity from the perspective of daily living disability resulting from cervical symptoms, primarily pain.
Ligament cell transfectionLigament cells were seeded in 6-well plates, and the siRNAs or plasmids were transfected at 50% confluence. Briefly, 2 μg of plasmid or siRNA (50 nmol/L) was mixed with 200 μL of jetPRIME buffer for 10 s. Then, 4 μL of jetPRIME transfection reagent (Polyplus, Cat# 0000001105) was added according to the manufacturer’s recommendation. After vortex and incubation, the mixture was added to antibiotics-free medium, and then the transfection medium was replaced by a growth medium 5 h later. We checked the transfection efficiency or conducted the following experiments after 36 h. The siRNAs sequences in this study are listed in Table S8.
Empty control plasmids (OE-Ctrl), wild-type LOXL2 overexpression plasmids (OE-LOXL2 WT), and point mutation LOXL2 plasmids (OE-LOXL2 Y689F) were procured from OriGENE Technologies, Inc.
Tube formation assay with ligament cellsAfter thawing at 4 °C overnight, we plated Matrigel (Corning, Cat# 356231) in 96-well culture plates and incubated at 37 °C to polymerize for at least 30 min. Transfected siRNA or plasmid ligament cells were digested by trypsin and resuspended in endothelial cell medium (ECM, 1001, ScienCell) supplemented with 5% FBS, 1% endothelial cell growth factors, and 1% penicillin/streptomycin. We then seeded (1 × 104 cells/well) on polymerized Matrigel. Sorafenib Tosylate (Cat# HY-10201A), Imatinib (Cat# HY-50946), recombinant human PDGF-BB (Cat# HY-P7055), VEGFA (Cat# HY-P7110A), VEGFA monoclonal antibody Bevacizumab (Cat# HY-P9906), and isotype control antibody (Cat# HY-P99001) were obtained from MedChemExpress (MCE). We observed and imaged the tube-like structures by microscopy after incubation at 37 °C for 4–6 h. The total length of tube-like structures was calculated with the help of ImageJ software (ImageJ; National Institutes of Health).
Trilineage differentiation of ligament cellsWe seeded ligament cells at 5 × 105 cells per well for osteogenic differentiation in the fibronectin-coated plate. The osteogenic differentiation medium (Cyagen Biosciences, Cat# HUXXC-90021) was changed every 2 days for differentiation. (2-Chloropyridin-4-yl)methanamine hydrochloride (Cat# HY-101771A) was acquired from MedChemExpress as a selective inhibitor of LOXL2 enzyme activity. ALP and Alizarin Red staining were used to testify to differentiation. The Alizarin Red was eluted by 10% cetylpyridinium chloride (wt/vol, Sigma-Aldrich, Cat# C9002). The absorbance of the extracted dye was measured at 570 nm. For adipogenesis differentiation, 8 × 105 cells were cultured in an adipogenesis differentiation medium (Cyagen Biosciences, Cat# HUXXC-90031) according to the manufacturer’s protocol for 14 days. Oil red staining was used to testify to differentiation. For chondrogenic differentiation, ligament cells were cultured in a chondrogenic differentiation medium (Sciencell, Cat# SC-7551) supplemented with 10 ng/mL recombinant human TGF-β3 (PeproTech, Cat# 100–36E) for 10 days, and the medium was changed every 2 days. Alcican blue staining was used to testify to differentiation.
Histochemical and Immunofluorescence analysisTo make paraffin-embedded tissue sections, we take the steps mentioned below. Firstly, fix the dissected tissue in 4% paraformaldehyde for 48 h. After rinsing in PBS, dissected specimens were decalcified in 0.5 mol/L EDTA pH 8.0 regent. Then the samples were dehydrated with ethanol from a concentration of 50%–100%. After soaking in xylene, the samples were transparent. Finally, paraffin was used to embed the tissue. The thickness of the sections was 6μm and prepared with a microtome (Leica RM2235) and mounted on slides. After baking at 62 °C for 1 h, slides were de-paraffined in xylene for 10 min twice. Then, a gradient of ethanol to water was used to rehydrate histological slides. Safranin O/Fast Green (SOFG)and Hematoxylin & Eosin (H&E) staining were conducted for histomorphometric analysis. Masson’s trichrome staining (Servicebio, Cat# G1006) and Sirius red staining (Solarbio, Cat# G1472) were performed according to the manufacturer’s instructions. The citrate antigen retrieval solution was heated to 95–97 °C for 10 min to perform antigen retrieval, and endogenous peroxidase activity was quenched by incubating samples with 3% hydrogen peroxide. Slides were permeabilized with 0.1% Triton X-100. The next steps followed the instructions of an immunohistochemistry Application Solutions Kit (CST, Cat#13079). A standard protocol was followed for immunofluorescence staining. Slides are de-paraffined and rehydrated following the same protocol as mentioned before. Citrate-EDTA antigen retrieval buffer was used 10–12 min after the temperature reached 95–100 °C. After being treated with 0.1% Triton X-100 and blocking, the primary antibody was incubated at 4 °C overnight. Primary antibodies were, anti-COLII (Abcam, Cat# ab34712, 1:100 dilution), anti-SP7 (Abcam, Cat# ab209484, 1:1 000 dilution), anti-BGLAP (SANTA CRUZ, Cat# sc-365797, 1:100 dilution), anti-MMP1(Abcam, Cat# ab52631, 1:100 dilution), anti-COLX (eBioscience, Cat# 14-9771-82, 1:100 dilution), anti-RUNX2 (SANTA CRUZ, Cat# sc-390351, 1:100 dilution), anti-CD31(Servicebio, Cat# GB11063-2-100, 1:200 dilution), specific anti-human CD31 antibody (Abcam, Cat# ab76533, 1:200 dilution), mouse anti-EMCN (Santa Cruz, Cat# sc-65495, 1:50 dilution), human anti-EMCN (Abcam, Cat# ab106100, 1 μg/mL), anti-LOXL2 (Abcam, Cat# ab96233, 1:500 dilution), anti-HIF-1 alpha (Abcam Cat# ab179483,1:500 dilution), anti-PRG4 (Abcam, Cat# ab28484, 1:200 dilution), anti-ACTA2(Abclonal,Cat# A17910, 1:100 dilution). We used Vector® TrueVIEW® Autofluorescence Quenching Kit (Vector Labs, Cat#: SP-8400-15) to minimize non-specific signals. After rinsing in PBS three times, the sections were incubated with secondary antibody for 1 h at room temperature. Slides were sealed with nail polish, and images were taken on the Olympus BX53 microscope or Leica SP8 confocal laser scanning microscope. Quantification was performed using ImageJ software (ImageJ; National Institutes of Health).
Western blottingThe cell lysates were prepared with RIPA buffer supplemented with protease/phosphatase inhibitor cocktail (MedChemExpress, Cat# HY-K0010) and 100 mol/L PMSF on ice. The relative protein concentration was measured using the BCA protein assay kit (Thermo-PIERCE, Cat# 23225). The protein lysates were separated using SDS-PAGE on 10% polyacrylamide gels, and molecular weight standards were determined with a prestained protein marker. The separated proteins were electro-transferred onto nitrocellulose filter (NC) membranes at ice-cold temperatures, followed by blocking in 5% bovine serum albumin for 1 h at room temperature. The appropriate primary antibodies, including anti-LOXL2 (Abcam Cat# ab96233, 1:1 000 dilution), anti-HIF-1 alpha (Abcam Cat# ab179483, 1:500 dilution), and anti-GAPDH (Abcam Cat# ab9485, 1:10 000 dilution), were rotated with the membrane at 4 °C overnight. Fluorescent secondary antibodies (LI-COR Biosciences, Cat# 926-32211, 1:10 000 dilution) were added for 2 h. Images were obtained using the LI-COR Odyssey CLx system, and densitometric analysis of protein bands was conducted using ImageJ.
Quantitative RT-PCRRNA was extracted with Trizol (Takara) to measure the relative mRNA levels. The absorbance ratio and concentration were determined by a NanoDrop 2000 (Thermo Fisher), followed by cDNA synthesis using RT reverse transcriptase kit (Hifair® II 1st Strand cDNA Synthesis SuperMix for qPCR (gDNA digester plus), Yeasen, Cat# 11123ES60) according to protocols. A QuantStudio 3 PCR system (Applied Biosystems/Thermo Fisher Scientific) was used to perform real-time quantitative analysis. The PCR program was 95 °C for 5 min, 40 cycles of following steps: 95 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s, and a total of 20 μL of reaction volume, including 10 μL of 2 × SYBR Master Mix(Hieff® qPCR SYBR Green Master Mix(Low Rox Plus), Yeasen, Cat# 11202ES08), 0.4 μL of each primer and 2 μL of diluted cDNA were used. For normalization, GAPDH (housekeeping gene) was used. Primers for amplification were listed in Table S9.
Transwell cell migration assaysTo evaluate the HUVEC (EA. hy926 cell line, Cat# GNHu39, Type Culture Collection of the Chinese Academy of Sciences) migration, we used medium supernatant of transfected siRNA or plasmid ligament cells to generate a conditioned medium. After 30 h of transfection, 4 mL medium supernatant was collected. The supernatant medium was centrifuged using a 3 kDa Ultra-4 Centrifugal Filter Unit (Millipore Amicon™, Cat# UFC800324) at 4 °C and 3 500 r/min for about 30 min to obtain 500 μL of concentrated medium supernatant and added to ECM medium at 1:8 dilution serving as lower chamber medium. HUVECs (2 × 104 cells/well) were seeded into the upper chamber (Falcon Cell Culture Inserts Cat# 353097, Corning) and inserted into the transwell. After 18 h, the upper chamber was fixed with 4% paraformaldehyde (wt/vol) and stained with crystal violet. We wiped off the non-migrated cells with a cotton swab, and the migrated cells were counted with the help of ImageJ (ImageJ; National Institutes of Health).
Flow cytometry analysisPrimary culture cells from non-OPLL or OPLL were digested with trypsin and washed in 2% FBS. We incubated cells for 30 min at 4 °C with primary antibody obtained from BioLegend at 1:200 dilution, PE anti-human CD45(Cat# 368509), PE anti-human CD73 (Cat# 344003), PE anti-human CD90 (Cat# 328109), FITC anti-human CD105 (Cat# 323203), ABflo® 594 anti-PDGFRA (Abclonal, Cat# A24332), ABflo® 488 anti-CD31/PECAM1 (Abclonal, Cat# A22508), ABflo® 647 anti-PROCR (Abclonal, Cat# A24227). After washing twice with 2% FBS, Fluorescence-activated cell sorting (FACS) was performed via flow cytometry (LSRFortessa™, BD Biosciences) and FACSDiva software (BD Biosciences). Flow cytometry data were analyzed using FlowJo v.10 software (FlowJo).
Lentiviral construction and transfectionFor LOXL2 overexpression, the LOXL2 coding sequence (CDS) was synthesized and cloned into HBLV-EF1-ZsGreen-PURO vectors. All lentiviral expression vectors were synthesized, packaged, and verified by HanBio Biotechnology (HanBio, Shanghai, China). Ligament cells were infected with different lentiviruses at a multiplicity of infection (MOI) of 30 in the presence of 5 µg/mL polybrene for 16–18 h.
Serum and medium supernatant VEGFA, PDGF-BB analysisWe used a VEGFA (R&D Systems, Cat# DVE00) and PDGF-BB Elisa kit (R&D Systems, Cat# DBB00) to determine the serum and medium supernatant VEGFA or PDGF-BB concentration, following the manufacturer’s instructions.
Micro-CT analysisCervical spines or ossified samples were scanned using x-ray microtomography (SKYSCAN 1272, Bruker Micro-CT, Belgium). The samples were scanned at a set of parameters (60.0 kV, 166.0 μA, and a resolution of 9-μm pixels). Two- and Three-dimensional images were reconstructed by Skyscan NRecon software (Bruker) and CTVox (Bruker Micro-CT), respectively. Bone parameters of the mice heterotopic ossification were calculated using CTan software (Bruker Micro-CT).
scRNA-seq using 10x genomicsObtaining ossified and non-ossified ligament tissue followed the steps described above. After thoroughly flushing the blood with saline, the collected samples were stored in MACS Tissue Storage Solution (Miltenyi, Cat# 130-100-008) at 4 °C. Subsequently, the harvested tissues were digested using RPMI 1640 medium (Gibco, Cat#11875093) supplemented with 0.3% type 1 collagenase (Gibco, Cat# 17100017) and 0.4% dispase II (Sigma-Aldrich, Cat# D4693) at 37 °C for 60 min under constant agitation. The digested tissues were then filtered through a series of strainers with pore sizes of 100 μm, 70 μm, and 40 μm. Following the tissue dissociation, the resulting cell suspension underwent treatment with red blood cell lysis buffer to eliminate red blood cells. Subsequently, the cell suspension was subjected to centrifugation, and the cell count was determined. The cells were resuspended at a concentration of 1 000 cells/μL. Due to current technological limitations, achieving a complete digestion of bone tissue to isolate its constituent cells was not feasible. Consequently, single-cell sequencing was conducted to examine cells from the PLL that displayed impending ossification and those from the non-ossified portion. Single-cell libraries were prepared using the Chromium Controller from 10x Genomics (Pleasanton, CA), following the protocol outlined in the Chromium Next GEM Single Cell 3’ Reagent Kits (v3.1, 10x Genomics). Libraries were prepared using the Chromium Controller from 10x Genomics. The final library pool was subjected to sequencing on the Illumina Novaseq6000 instrument, generating 150 base-pair paired-end reads. Alignment to the GRCh38 reference genome, quantification of gene expression, and aggregation of sample count matrices were performed using the cellranger pipeline version 6.1.1.
Bioinformatics analysis of single-cell sequencing dataData set integration, dimensionality reduction, and clustering were conducted with Seurat version 4.3.0. Cells meeting the criteria of nFeature_RNA > 200, nFeature_RNA < 5 000, and <15% mitochondrial genes were subjected to further processing. Sctransform package version 0.3.5 and harmony package version 0.1.1 were employed to address batch effects. UMAP (Uniform Manifold Approximation and Projection) dimensionality reduction was applied using the top 50 components. For initial clustering, 40 dimensions of reduction were used as input, and the Louvain algorithm with a resolution of 0.2 was employed through the Findcluster function. Among the clusters, two displayed high hemoglobin gene expression, indicating potential red blood cell contamination. Consequently, these cells were removed. To perform sub-clustering of ligament cells, pericytes, and endothelial cells, dimensionality reduction and clustering were carried out using the Monocle3 package version 1.3.1. Eight subclusters were identified using a resolution of 3e-5. Single-cell trajectory analysis was performed on the cell clusters using the Monocle3 package. UMAP was used for dimensionality reduction, and the learn_graph function in Monocle3 was employed for pseudotime trajectory construction, with the C6_progenitor designated as the root node. The identification of the root node was facilitated by the CytoTrace package version 0.3.3 based on cytoTrace scores and velocyto (version 0.2.5) based on RNA velocity. Furthermore, the find_gene_module function was utilized to group co-regulated genes into modules using Louvain community analysis. Module scores were calculated by aggregating the expression of all genes within each module across all clusters and were visualized using heatmaps. Functional enrichment analysis of each module was performed using the ReactomePA package version 1.42.0.
Transcriptome sequencingThe total RNA was extracted with TRIzol reagent (Invitrogen). The RNA amount and purity quantification, RNA integrity number (RIN) assessment, library preparations, and paired-end sequencing were performed by Origin-gene Bio-Pharm on a novaseq6000 platform (Illumina).
Protein-protein interaction (PPI) network constructionAll the DEGs identified in the tissue transcriptomic sequencing were uploaded to an online database - STRING, and analyzed with default parameters. The tsv format files were downloaded and imported into Cytoscape software (Version: 3.8.2). Cytoscape plugin cytoHubba for network centrality analysis was used to identify a network’s critical nodes (genes). Our study used 5 specific centrality measures to identify critical genes (hub genes) by calculated scores. The methods were listed: Maximal Clique Centrality (MCC), Density of Maximum Neighborhood Component (DMNC), Edge Percolated Component (EPC), Maximum Neighborhood Component (MNC), and ClusteringCoefficient.
Bioinformatics analysisFirstly, adapter contamination, low-quality bases, and undetermined bases were removed by trim_galore version 0.6.6, and sequence quality was verified using FastQC v0.11.9. We used HISAT2 version 2.2.1 and samtools version 1.7 to map filtered reads to the human genome GRCh38.p13 in Ensemble104. After the final transcriptome was generated, featureCounts version 2.0.1 was used to obtain strand-specific read counts. The Differentially expressed genes (DEGs) were selected with |log2 (fold change)| > 1 and with statistical significance (P < 0.05) by R package edgeR. Then clusterProfiler was used for Gene Ontology (GO) functional annotation analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis, and Gene set enrichment analysis (GSEA).
AnimalsFor the BMP-induced in vivo ossification (BIO) model, male C57BL/6J mice (6 weeks old) were obtained from Shanghai Leigen Biotechnology Co., Ltd. After being anaesthetized with an intraperitoneal injection of Avertin, a longitudinal incision of ~1 cm was made on the back of each mouse. Then, an rhBMP-2 biomaterial (Hangzhou Jiuyuan Gene Engineering Co., Ltd., China) containing gelatin, soy lecithin, and hydroxyapatite was implanted under the skin and secured to the back muscles with a 4–0 absorbable collagen thread. The surgical incisions were closed with 5–0 nylon sutures. The mice were housed in a laminar airflow cabinet with specific pathogen-free conditions, a room temperature of 22 °C, and a 12-h light/dark cycle. On the second postoperative day, sorafenib was resuspended in 0.5% sodium carboxymethylcellulose at a concentration of 50 mg/kg body weight. The intervention mice were administered sorafenib by gavage once a day for 14 days, while the vehicle group was given the same volume of sodium carboxymethylcellulose by intragastric administration. The mice were sacrificed by excessive anesthesia, and the heterotopic ossification on the back was excised for subsequent analysis.
For Ligament Spontaneous Ossification (LSO), enpp1 KO mice (strain NO.T013898) were obtained from GemPharmatech (Nanjing, China). Four-week-old enpp1 KO mice were used in this study, and sorafenib was administered orally to the mice daily. The cervical spines were collected at 18 weeks. The raising, administration, and sample collection strategies were the same as those described in the BIO model. The genotypes of transgenic mice were determined by PCR (Absin, Cat# ABS60036) using genomic DNA isolated from mice tails. The primer pairs are listed in Table S10.
In vivo Matrigel plug assayNude mice aged 6–8 weeks were anaesthetized with Avertin. The back skin of the nude mice was gently lifted, and 0.2 mL of cold Matrigel, with or without 2 × 106 ligament cells, was injected into the subcutaneous right and left dorsolateral sides to create hillock-like and pushable bulges. After 7 days, the mice were anaesthetized and perfused sequentially with heparinized saline, 4% PFA, and ink (Kuretake, Cat# CE100-6) through the left ventricle. Subsequently, the previously injected Matrigel was removed once the nude mice had utterly darkened. The ink particle deposits in lumen-like structures on sections indicated the formation of functional blood vessels. The dyes Ulex Europaeus Agglutinin I (UEA I, Cat#DL-1067) and Griffonia Simplicifolia Lectin I Isolectin B4 (GS-B4, Cat#DL-1208) used for fluorescence staining were purchased from Vector Laboratories.
Evaluation of the sensory and motor function of miceTo evaluate the effects of sorafenib on mice’s sensory and motor function, mechanical and thermal nociception hyperalgesia tests were adopted. Research blinded to the study groups used electronic von Fray filaments (BIO-EVF4, Bioseb, France) to evaluate mechanical sensitivity and plantar test apparatus (Ugo-Basile 37370, Comerio, Italy) to perform thermal hyperalgesia measurement. Following our previous protocol, mice were placed in cages or on the glass plate for 30 min before the test started. Three experimental data were recorded as manufacturer’s instructions when the hind paws began to buckle for the mechanical nociception hyperalgesia test. For the thermal hyperalgesia test, we recorded the time from irradiation to mouse hind paw withdrawal, at least a 30-min interval between trials. Three experimental data of each mouse were recorded as response times.
Statistical analysisData were presented as mean value ± SD (standard deviation) except for some baseline characteristics, which were reported as counts and percentages for categorical variables or median and interquartile range (IQR) when normality assumptions were not met. Student’s t-test or One-way ANOVA with Tukey’s multiple comparisons was performed as appropriate, while the Mann-Whitney U test or Kruskal-Wallis test was used for non-normally distributed data. Pearson correlation was used to assess the correlation between two variables. The diagnostic performance of serum VEGFA for OPLL was determined using a ROC curve and the area under the ROC curve (AUC). Baseline characteristics were presented as mean ± SD for continuous variables, counts, and percentages for categorical variables. Student’s t-test and chi-squared tests or Fisher’s Exact Test were used to compare continuous and categorical characteristics, respectively, between non-OPLL and OPLL. Duration of symptoms was tested using covariance analysis with a univariate general linear model, adjusting for age. All statistical analyses were performed using SPSS 23.0 (IBM, Chicago) and GraphPad Prism 8.0 (GraphPad, San Diego). Statistical significance was considered at P < 0.05 (*P < 0.05, **P < 0.01, ***P < 0.001), while ns indicated non-significance. At least three independent experimental groups were analyzed.
留言 (0)