
記住我

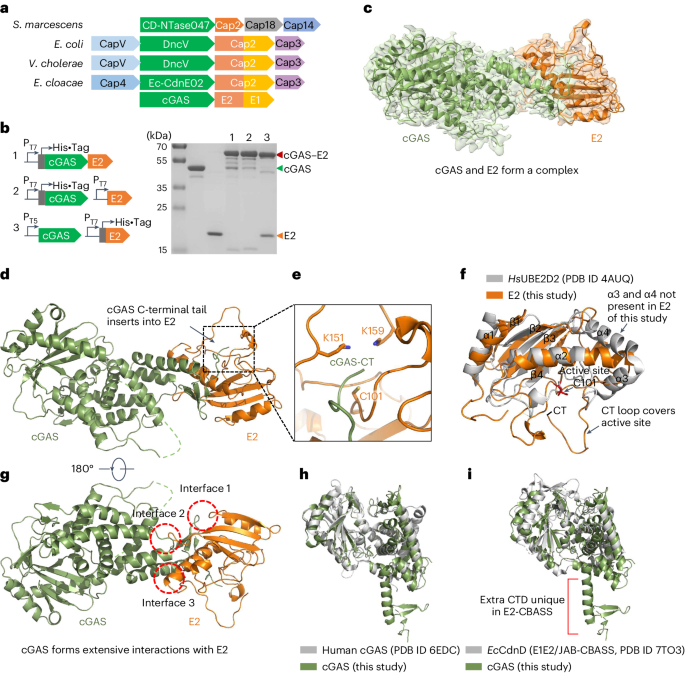

E. coli DH5α and BL21 (DE3) or C43 (ref. 40) were used for cloning and for the expression of recombinant proteins, respectively. All B. subtilis strains used in this study are derivatives of the laboratory strain 168. B. subtilis and E. coli were grown in Lysogeny Broth (LB) or in sporulation (SP) medium40,41. For growth assays and the in vivo interaction experiments, B. subtilis was cultivated in LB. The media were supplemented with ampicillin (100 µg ml−1 for E. coli), kanamycin (50 µg ml−1 for E. coli, 10 µg ml−1 for B. subtilis), chloramphenicol (35 µg ml−1 for E. coli, 5 µg ml−1 for B. subtilis), erythromycin and lincomycin (2 and 25 µg ml−1, respectively), or Zeocin (35 µg ml−1) if required.
Isolation of novel B. subtilis phagesThe laboratory strain B. subtilis Δ6 (ref. 23), a derivate of B. subtilis 168, was used as host for the isolation of novel phages as described previously42. Briefly, raw sewage from a municipal sewage treatment plant in Göttingen, Germany served as environmental phage source and was cleared by centrifugation at 5,000 g for 10 min, following filtration through a sterile filter (0.45 μm, Sarstedt). The host was infected by mixing 100 μl of an overnight culture with 2 ml of processed sewage water, followed by 5 min incubation at room temperature (r.t.) to allow adsorption. This bacterial suspension was then mixed with 2.5 ml of 0.4% agarose (50 °C) (Fisher BioReagents) dissolved in LB medium, spread on a LB plate and incubated overnight at 28 °C. Individual plaques were picked, serially diluted and higher dilutions were used for re-infection of the host. Cells were grown until complete lysis and the supernatant processed as described above. This re-infection was repeated to scale up the phage lysate. For genome sequencing of the novel isolates, phage DNA was isolated as previously described43. Sequencing was performed using an Illumina MiSeq sequencer (Illumina). Phages were classified according to nucleotide sequence homology to the closest relative as revealed by Megablast in the Geneious Prime 2022.2.1 software. The genomic sequences of the new phages and their closest relative (downloaded from NCBI on 10 March 2023) were aligned using MMseqs in the pyGenomeViz package (https://github.com/moshi4/pyGenomeViz, accessed on 10 April 2023; input: gbk files; mode: pgv-mmseqs). A list of all phages isolated in this study can be found in Supplementary Table 1.
Phage amplification and storagePhage lysates were generated from B. subtilis Δ6 using the modified double agar overlay plate technique. For this, a culture of B. subtilis Δ6 was precultured to late exponential growth phase in LB medium. Bacterial cells (100 µl) were mixed with 100 µl phage dilution, incubated for 5 min at r.t. to allow adsorption of the phage, mixed with 2.5 ml LB supplemented with 0.4% agarose (50 °C) and poured over a prewarmed (37 °C) LB plate. Plates were incubated overnight at 37 °C. Phages were collected the following day by adding 5 ml of LB medium directly onto the plate, incubating for at least 30 min at r.t., and collecting and filtering the resulting liquid through a 0.2 µm Nanosep filter. Lysate titres were determined by infection of B. subtilis Δ6 using the modified double agar overlay plate technique as described above and the resulting p.f.u. ml−1 was calculated. Phage lysates were stored at 4 °C.
Phage infection assaysPhage infection assays were performed on plates and in liquid culture. The desired B. subtilis strains were precultured overnight at 28 °C and used to inoculate fresh LB medium. This culture was grown until late exponential growth phase. For infection on plates, 100 µl were mixed with 2.5 ml LB supplemented with 0.4% agarose (50 °C) and poured over a prewarmed LB plate. Serial dilutions of the phage lysate were spotted onto this plate. Plates were incubated overnight at 37 °C and pictures were taken the following day. For quantification, p.f.u. ml−1 was calculated from at least three biological replicates. Fold defence was calculated as the ratio between the p.f.u. ml−1 obtained from wild type (no CBASS) and strains containing the defence system. For infection in liquid medium, the optical density of the culture at 600 nm (OD600) was adjusted to 1.0. This culture was then used to inoculate fresh LB medium in a 96-well plate (Microtest Plate 96-well, Sarstedt) to OD600 of 0.1 (1:10 dilution). Growth was tracked in an Epoch 2 microplate spectrophotometer (BioTek Instruments) at 37 °C with linear shaking at 237 cpm (4 mm) for 20 h, and OD600 was measured at 2 min intervals.
DNA manipulationTransformation of E. coli and plasmid DNA extraction were performed using standard procedures40. All commercially available plasmids, restriction enzymes, T4 DNA ligase and DNA polymerases were used as recommended by the manufacturers. Chromosomal DNA of B. subtilis was isolated as previously described36. B. subtilis was transformed with plasmid and genomic DNA according to the two-step protocol36.
Construction of mutant strains by allelic replacementDeletion of the pspA gene was achieved by transformation of B. subtilis Δ6 with a PCR product constructed using oligonucleotides to amplify DNA fragments flanking the target genes and an appropriate intervening resistance cassette as previously described44. The integrity of the regions flanking the integrated resistance cassette was verified by sequencing PCR products of ~1,100 bp amplified from chromosomal DNA of the resulting mutant strains. A list of all strains constructed in this study can be found in Supplementary Table 2.
Plasmid construction and mutagenesisThe wild-type CBASS operon from B. cereus WPysSW2 was ordered as genomic DNA from Integrated DNA Technologies (IDT) and the desired genes were amplified using appropriate oligonucleotides that attached specific restriction sites to the fragment. Plasmids derived from pGP1460 (integration into lacA)45 were linearized with ScaI for genomic integration. The integrity of the integration was confirmed by PCR and subsequent sequencing of the region.
For protein expression in E. coli, the genes encoding CD-NTase, Cap2, Cap3, Nuc-SAVED and PspABce were codon optimized and obtained from IDT. Each gene was subcloned into the vector pEhisV5TEV46, allowing expression with a cleavable N-terminal polyhistidine tag, or into pGP172 for expression with an N-terminal Strep-tag. The pspABsu gene was amplified from B. subtilis 168 chromosomal DNA using oligonucleotides that attached specific restriction sites to the fragment and cloned into the vector pEhisV5TEV. The PspA variants K147 and K220 were generated by site-directed mutagenesis of the pspA-pEhisV5TEV vector. For co-expressions in E. coli, the cyclase gene remained within the pEhisV5TEV vector to retain cleavable polyhistidine tag, while other proteins (Cap2, Cap3 and PspABsu) were expressed on pCDFduet and pACYCduet. All synthetic genes, plasmids and primers are listed in Supplementary Tables 3–5.
Protein expression and purificationE. coli C43 (DE3) was transformed with the plasmid pEhisV5TEV encoding the wild-type Bce cyclase or a variant truncated after amino acid 301, Nuc-SAVED, Cap2, Cap3, wild-type PspA, or the variants, or pGP172 encoding Strep-Cap2. Expression of the recombinant proteins was induced by the addition of isopropyl 1-thio-β-d-galactopyranoside (final concentration, 0.4 mM for pEhisV5TEV or 1 mM for pGP172) to exponentially growing cultures (OD600 of 0.8) of E. coli carrying the relevant plasmid. The Bce cyclase and Cap3 were induced overnight at 25 °C. The truncated cyclase was induced for 4 h at 25 °C. The Nuc-SAVED protein and Strep-Cap2 were induced overnight at 16 °C. Cell pellets were resuspended in lysis buffer (50 mM Tris-HCl, 250 mM NaCl, 10 mM imidazole, 10% glycerol, pH 7.5) supplemented with 1 mg ml−1 lysozyme and protease inhibitor (Roche). The cells were lysed by sonication on ice six times for 1 min at an amplitude of 12, with 1 min pause on ice in between (MSE Soniprep 150). After lysis, the crude extract was centrifuged at 117,734 × g for 30 min and then passed over a Ni2+nitrilotriacetic acid column (His-trap FF crude, Cytiva). Ni2+nitrilotriacetic acid columns were washed with wash buffer (50 mM Tris-HCl, 250 mM NaCl, 30 mM imidazole, 10% glycerol, pH 7.5) to clear unbound protein. Target protein was eluted with an imidazole gradient or biotin (5 mM). After elution, the fractions were tested for the desired protein using SDS–PAGE. To remove the 8x-His-TEV tag from the proteins, the relevant fractions were combined and the tag was removed with the TEV protease (ratio 10:1 w/w) during overnight dialysis (Biodesign Cellulose Dialysis Tubing Roll, 10 kDa) against wash buffer. The cleaved TEV moiety and the protease were removed using a fresh Ni2+nitrilotriacetic acid column. The purified protein was concentrated in a Merck Amicon Ultra-15 Centrifugal Filter device (cut-off 10 kDa). The protein was loaded on a HiLoad 16/600 Superdex 200 pg column pre-equilibrated with gel filtration buffer (20 mM Tris-HCl, 250 mM NaCl, 10% glycerol, pH 7.5) and the fractions containing pure protein were collected and concentrated again. The protein samples were stored at −70 °C until further use.
Expression of wild-type PspABsu or the protein variants and PspABce was induced with 0.4 mM isopropyl-β-d-1-thiogalactoside (IPTG) and cells grown for 4 h at 25 °C. PspA was purified as described above, except that the Ni2+nitrilotriacetic acid column was washed with 20 column volumes (CV) of wash buffer (containing 50 mM Tris-HCl pH 7.5, 500 mM NaCl, 30 mM imidazole and 10% glycerol), then 4 CV of 50 mM Tris-HCl pH 7.5, 500 mM NaCl, 50 mM imidazole and 10% glycerol, and then eluted directly with 6 CV of elution buffer (containing 50 mM Tris-HCl pH 7.5, 500 mM NaCl, 500 mM imidazole and 10% glycerol). The eluted peak was pooled and concentrated using an Amicon Ultra-15 (Millipore) centrifugal filter (MW cut-off 10 kDa) before loading onto an equilibrated HiTrap desalting column (Cytiva), then washed with 50 mM Tris-HCl pH 7.5, 500 mM NaCl, 30 mM imidazole and 10% glycerol. The desalted protein was collected and treated with TEV protease for 2 h at r.t. with gentle agitation. The protein was isolated from TEV protease by passing through another His-Trap FF crude column, and the unbound fraction collected and concentrated. PspA was further purified by SEC (S200 16/60, Cytiva) in buffer containing 20 mM Tris pH 7.5, 500 mM NaCl and 10% glycerol. Chosen fractions were concentrated and aliquoted, then frozen at −70 °C.
Strep-tagged proteins were expressed, lysed and ultracentrifuged as described for His-tagged proteins above in buffer W (100 mM Tris-HCl, 150 mM NaCl, pH 8.0), unless stated otherwise. The crude extract was then applied to a StrepTactin column (IBA Lifesciences) and the column washed with 100 mM Tris-HCl and 150 mM NaCl pH 8.0. The protein was eluted with a biotin gradient (5 mM). Relevant fractions were identified by SDS–PAGE and concentrated using Vivaspin turbo 15 (Sartorius) before storage as described above.
Co-expression and purification of the cyclaseBce–PspABsu conjugateCo-expression strains (His-tagged cyclaseBce, untagged PspABsu and Cap2Bce) were generated by transformation of E. coli C43 (DE3) with pV5SpHISTEV containing wild-type cyclaseBce or the truncated variant (1–301), in addition to the dual-expression vector pCDFduet that contained PspABsu and Cap2Bce. Cultures were induced for protein expression with 0.4 mM IPTG for 4 h at 25 °C, conditions previously identified as optimum for cyclase–PspA conjugate formation46. Cyclase–PspA conjugates were purified following the His-tagged protein method detailed above. As the only protein with His-tag present was the cyclase, minimal Cap2/PspA were expected unless conjugated to/interacting with cyclase. Multiple cyclase–PspA conjugation products were separated through SEC on a HiLoad 26/600 Superdex 200 pg column, at a flow rate of 1.5 ml min−1 for increased separation of varying-MW species. Fractions containing individual conjugation species were then identified via SDS–PAGE and pooled separately before concentrating and storing at −70 °C as above.
Pulldown of cyclaseBce in E. coliCo-expression strains of the His-cyclase and PspA with or without additional expression of Cap2 or Cap3 were grown in 10 ml LB and induced during exponential growth phase with 0.4 mM IPTG. Expression strains were then incubated for 4 h at 25 °C and cells collected by centrifugation. His-tagged cyclase was purified and visualized on SDS–PAGE as previously described46. Briefly, cell pellets were resuspended in lysis buffer (50 mM Tris-HCl, 250 mM NaCl, 10 mM imidazole, 10% glycerol, pH 7.5) before being lysed by sonication on ice. The cell lysate was then centrifuged at 10,000 × g for 10 min before supernatant was added to Biosprint plasticware. His-tagged proteins were isolated using Nickel beads (His Mag Sepharose Ni, GE Healthcare) in the QIAGEN BioSprint machine, with wash and elution buffers containing 50 mM Tris-HCl, 500 mM NaCl, 10% glycerol (pH 7.5) and 30/500 mM imidazole, respectively. Eluted His-tagged protein and total protein-containing fractions were analysed by SDS–PAGE and western blot.
Purification of cyclaseBce from B. subtilis and separation of cell extractFor overexpression of the cyclase in B. subtilis, strain LK40 was transformed with either pLK20 (Strep-cyclase), pLK21 (Strep-cyclaseΔC) or the empty vector control (pGP380). For comparison of the levels of cyclase conjugate, the B. subtilis strains LK06, LK10, LK18, LK20 and LK51 containing single genomic integrations of the CBASS operon were used. The respective strains were cultivated in LB medium until exponential growth phase was reached (OD600~0.5). If desired, cells were infected with the phage Goe23 and incubated for another 5, 15 or 30 min. The cells were collected by centrifugation at 3,220 g and 4 °C, and the pellets stored at −20 °C. For separation of cytosolic and membrane fractions of the crude extract, the pellets were resuspended in buffer M (50 mM NaH2PO4, 50 mM Na2HPO4, pH 6.8) supplemented with 1 mg ml−1 lysozyme and protease inhibitor (Roche), and opened by sonication. To remove cell debris and unbroken cells, the lysate was centrifuged at 3,220 g for 20 min at 4 °C. The supernatant was subjected to ultracentrifugation at 21,000 g for 90 min at 4 °C to pellet the membranes. The supernatant containing cytosolic proteins was removed, the pellet washed with buffer M and the membranes pelleted again in an additional centrifugation step (21,000 g, 60 min, 4 °C). The supernatant was discarded and the wash step repeated one more time. The resulting pellet containing the membranes was resuspended in buffer M. To achieve solubilization of the membranes, the fraction was 1:1 diluted with buffer M containing 0.5% n-Dodecyl-β-D-maltoside ()(final concentration of 0.25%) and incubated overnight with rotation at 4 °C. The protein concentration of cytosolic and membrane fractions was determined, and equal amounts of the fractions were separated on an SDS gel and analysed by staining with Coomassie blue and western blot analysis. To confirm the proper separation of cytosolic and membrane fractions, membranes were also developed with α-HPr and α-Rny primary antibodies as cytosolic and membrane protein controls, respectively. The cyclase was further purified from both cytosolic and membrane fractions or the entire cell lysate using Protein A-coupled Dynabeads (Invitrogen) that were saturated with α-cyclase antibody for immunoprecipitation. The eluates were separated by SDS–PAGE and analysed by western blot. Bands of interest were subjected to further analysis by MS. For purification of the cyclase from cell lysates without separation into cytosolic and membrane fractions, the pellets were resuspended in buffer W (100 mM Tris-HCl, 150 mM NaCl, pH 8.0) supplemented with 1 mg ml−1 lysozyme and protease inhibitor (Roche), and opened by sonication. To remove cell debris and unbroken cells, the lysate was centrifuged at 20,000 g for 10 min at 4 °C. Protein concentration of the lysates was determined and adjusted with buffer W. The cyclase was purified from the lysates using Protein A-coupled Dynabeads (Invitrogen) saturated with α-cyclase antibody. The eluates were separated by SDS–PAGE and analysed by western blot.
Western blot analysisPolyclonal antibodies against the B. cereus cyclase or the PspA protein from B. subtilis were produced in rabbits (by Kaneka Eurogentec), and the anti-cyclase antibody was purified with Cyanogen bromide-activated Sepharose (Merck) by immobilizing purified cyclase. The purified α-cyclase antibody was used at 1:10,000 dilution for detection and the α-PspABsu serum at 1:5,000 dilution. The α-HPr antibody was used at 1:10,000 dilution and the α-Rny antibody at 1:100,000 dilution. Western blot analysis was performed on cell extracts and purified protein to follow the modification of the cyclase protein. Samples were resolved using SDS–PAGE and transferred to PVDF membranes. Membranes were blocked for at least 1 h at r.t. in blocking buffer (1× PBS, 5% w/v milk powder, 0.025% Tween20). Incubation with the respective antibodies (α-cyclase, α-HPr, α-Rny) was carried out overnight at 4 °C. Blots were extensively washed and incubated with the α-rabbit secondary antibodies (IRDye 800CW donkey anti-rabbit IgG secondary antibody; LI-COR) at 1:20,000 dilution in blocking buffer. The specific protein bands were visualized using the LI-COR Odyssey CLx and images adjusted using the ImageJ software (http://rsb.info.nih.gov/ij/index.html). Bands were analysed and quantified with ImageJ by comparing the intensity of selected bands to a standard band on the same blot. Data were analysed and visualized using GraphPad Prism 9.5.1 (Dotmatics).
Conjugation assayFor reconstruction of the conjugation in vitro, the cyclase (5 µM) was incubated together with Cap2 (5 µM), PspA (5/10 µM), 10 mM MgCl2 and 1 mM ATP in 100 mM Tris-HCl and 150 mM NaCl (pH 8.0) for 30 min at 25 °C. The samples were separated by SDS–PAGE and analysed by Coomassie staining or western blot with an antibody raised against the cyclase.
Mass spectrometry analysisProtein bands were excised from the gel and prepared for MS analysis using established protocols47. Briefly, this included destaining with ethanol:water, reduction with dithiorethritol and alkylation with iodoacetamide. Samples were digested with either trypsin, AspN or a combination of trypsin and GluC. The peptides from the gel pieces were soaked and the eluent concentrated to 20 µl. The samples were analysed by nanoLC–MS/MS on a ThermoScientific Fusion Lumos Orbitrap mass spectrometer coupled to a ThermoScientific u3000 nanoLC. An LC was configured in trap elute format, with the Acclaim Pepmap 100 (100 µm × 2 cm) nanoViper trap and the Pepmap RSLC C18 3 µm 100A (75 µm × 15 cm) Easyspray column (both ThermoScientific). Of the sample, 5–10 µl was injected onto the trap at 15 µl min−1 of loading buffer (0.05% trifluoroacetic acid in water) and run for three min. The trap was switched in line with the analytical column and the sample eluted at a gradient over 65 min (A = 100% water with 0.1% formic acid, B = 20% water 80% acetonitrile, 0.1% formic acid, 2% A to 3 min, linear to 40% A over 42 min, linear to 95% A over 4 min, hold for 5 min, linear back to 2% A and re-equilibrate for 10 min). The flow from the column was sprayed directly into the Easyspray orifice at a voltage of 1,700 V positive ionization. MS data were collected from 350 to 2,000 on the orbitrap at a resolution of 120,000 for the survey scan and a cycle time of 2 s for data-dependant acquisition conditions for MSMS on the orbitrap trap at a resolution of 30,000. Both electron transfer dissociation (ETD) and higher energy collison dissociation (HCD) fragmentation techniques were used. Raw data were exported and extracted using ms convert (ProteoWizard). The data were searched using the Mascot search engine (MatrixScience) against an internally generated database of 6,700 protein sequences containing the sequences of our recombinant proteins or the B. subtilis specific protein database (UniProt Proteome ID UP000001570). Settings were 20 ppm on the MS and 0.1 Da on the MSMS data, with a fixed modification of carbamidomethyl on cysteine and variable oxidation of methionine. For the identification of the conjugation site on PspA, the mass of the last C-terminal tryptic peptide of the cyclase (KPGGFA) minus one water molecule (C(27) H(39) N(7) O(6) 557.296182) was set as a variable modification on lysine residues. Peptides identified as modified are shown in Supplementary Table 6.
Intact mass measurement was carried out on a Waters Xevo G2S TOF mass spectrometer with Waters Acquity LC. A volume of 10 µl of 1 µM sample was desalted online through a Waters MassPrep On-Line Desalting Cartridge (2.1 × 10 mm), eluting at 200 µl min−1 with an increasing acetonitrile concentration (2% acetonitrile, 98% aqueous 1% formic acid to 98% acetonitrile, 2% aqueous 1% formic acid) and eluted directly into the MS with a lock mass of LeuEnk to ensure stable calibration. The spectra across the elution peak were combined and the charged ion envelope deconvoluted with MaxEnt algorithm using the peak width at half height of the most intense peak, to a resolution of 0.1 Da.
Cyclase product quantification by HPLCTo obtain cyclase products, the reaction was set up as follows: 50 µM cyclase, 0.2 mM/5 mM ATP in cyclase buffer (50 mM HEPES, 150 mM KCl, 10 mM MgCl2, 10% glycerol, pH 7.5) for 20 min/45 min. Reaction samples were filtered through a Pall Nanosep spin filter (3 kDa cut-off) by centrifugation at 10,000 g for 10 min to remove protein. A volume of 5 µl of product or synthetic standard was injected onto a C18 column (Kinetex EVO P 2.1 ×50 mm, particle size of 2.6 µm) attached to a Thermo UltiMate 3000 chromatography system. Absorbance was monitored at 260 nm and 40 °C. Gradient elution was performed with solvent A (100 mM ammonium acetate) and solvent B (100% methanol plus 0.1% trifluoroacetic acid) with a flow rate of 0.3 ml min−1 as follows: 0–0.5 min, 0% B; 0.5–3.5 min, 20% B; 3.5–5 min, 50% B; 5–10 min, 100% B. The area under the peak (mAU (milli-absorbance units) × min) was determined with the Chromeleon 6.80 software (Dionex). The activity of the cyclase was calculated by fitting the area under the peak values to those of a cA3 standard curve. Data were analysed and visualized using GraphPad Prism 9.5.1 (Dotmatics).
Cyclase product quantification by fluorescent Nuc-SAVED assayWild-type cyclaseBce, the truncated variant (1–301) or the cyclaseBce–PspABsu conjugate were incubated with ATP under standardized conditions for comparison of product formation. Cyclase (20 µM) was incubated with 250 µM ATP in cyclase buffer (50 mM HEPES, 150 mM KCl, 10 mM MgCl2, 10% glycerol, pH 7.5) for 1 h, unless stated otherwise. Reactions were quenched with 10 mM EDTA and cyclase denatured at 95 °C for 5 min. For rapid quantification of the relative cA3 produced by the cyclase variants, a dilution of the cyclase reaction was incubated with the CBASS effector Nuc-SAVED. Method and conditions were adapted from an analogous previously described NucC assay31. The assay contained 50 mM Tris-HCl (pH 7.5), 20 mM NaCl, 10 mM MgCl2, 10% (v/v) glycerol, 100 nM FAM:Iowa Black double-stranded DNA substrate, 100 nM Nuc-SAVED and 500× dilution of denatured cyclase reaction or 25 nM synthetic cA3, unless otherwise stated. The absorbance was measured with a FluoStar Omega plate reader (BMG Labtech) using fluorescence detection (ex/em 485/520 nm) in black non-binding half-area 96-well plates (Corning). Fluorescence was measured at 30 s intervals at 37 °C. The reaction was initially incubated in the absence of cA3/cyclase product for 10 min to measure a baseline. The measurement was stopped, synthetic cA3/cyclase product added and the measurement continued for 50 min. A standard curve was generated for Nuc-SAVED with varying synthetic 3′5′ cA3 concentrations (0.1 nM–1 µM) and the initial rate of reaction used to generate a standard curve of rate vs [cA3]. For the comparison of the different cyclase variants, the cyclase reaction sample was analysed by SDS–PAGE additionally to normalize the protein amount to the initial rate, as the conjugate sample was less pure than the wild type and the truncated cyclase. Bands were analysed and quantified with ImageJ by comparing the intensities of selected bands on the same gel. Data were analysed and visualized using GraphPad Prism 9.5.1 (Dotmatics).
Extraction of nucleotidesFor the extraction of nucleotides, B. subtilis cultures carrying genomic integrations of the respective CBASS version were grown until exponential growth phase at 37 °C. If indicated, the cultures were infected with phage Goe21, Goe23 or Goe26 to a multiplicity of infection (MOI) of 2 and grown for additional time as indicated. A volume of 50 ml of the cultures was collected immediately and the pellets resuspended in extraction solution (methanol:water:acetonitrile, 2:2:1). The cells were lysed and the proteins denatured by vortexing for 20 s and incubation at 95 °C for 10 min. The samples were stored at −70 °C overnight. After thawing, the denatured proteins were separated by centrifugation at 21,000 g and the protein pellet used to determine the protein amount in each sample. The supernatants containing the cell-free extracts were dried at 40 °C in a Speed-Vac. The dried pellets were resuspended in 60 µl of water and filtered through a Pall Nanosep spin filter (3 kDa cut-off) by centrifugation at 10,000 g for 10 min. The filtrate was tested for the CBASS-specific nucleotide in a plasmid cleavage assay.
Plasmid cleavage assayThe assay contained 20 mM HEPES (pH 7.5), 10 mM NaCl, 10 mM MgCl2, 10% (v/v) glycerol, 75 nM plasmid DNA, 1 µM CBASS effector protein Nuc-SAVED and 1.5 µl of the extracted nucleotides or the diluted cyclase reaction product (1:1,000/1:10,000) per 15 µl reaction sample or synthetic cA3, unless stated otherwise. The reaction was started by the addition of the nucleotide extracts or the synthetic standard, and the samples incubated at 37 °C for 30 min, or 10 min in the case of the cyclase reaction products or, in the case of the time course experiment, for the stated amount of time. The samples were mixed with DNA loading dye (TriTrack, ThermoScientific) and separated on a 1% agarose gel (run in 1× TBE buffer). The DNA was stained with SYBR-Green (1:50,000, in 1× TBE) and the gels imaged on a Typhoon FLA 7000 fluorescence imaging system (Cytiva). The band of the undigested supercoiled plasmid was quantified with ImageJ by comparing the intensity of selected bands to a standard band on the same gel. If indicated, relative values were normalized to the amount of protein extracted in parallel to the nucleotides from the same cultures. Data were analysed and visualized using GraphPad Prism 9.5.1 (Dotmatics).
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
留言 (0)