
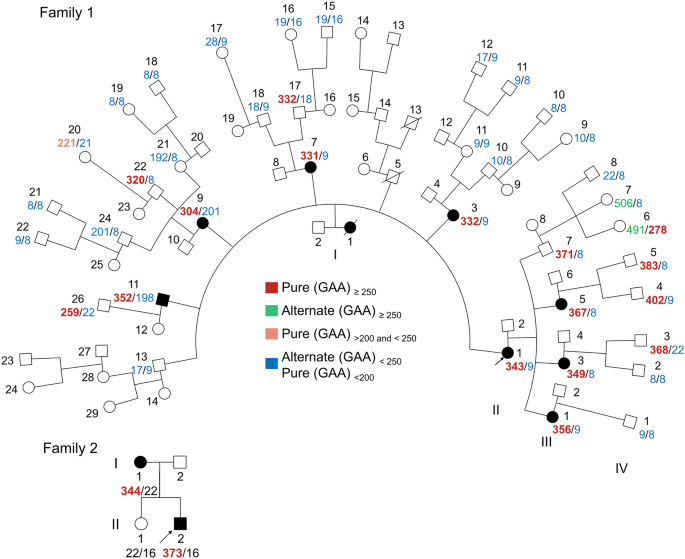
記住我
Our evolutionary framework regards selection on a complex trait considered to have been under selection in the past. Whilst this selection is strong, it is distributed over many loci leading to “nearly neutral” behaviour for each locus under a range of conditions (e.g. Eyre-Walker [24]). This facilitates a neutral model by assuming that selection has maintained SNPs at or below the threshold where selection would efficiently reduce their frequency [19]. This model is practical for inference and widely used for inference of heritability (e.g. Yang et al. [25]), and is appropriate following a contraction if selection is either negligible (because it is less effective in a smaller or growing population) or absent (because the conditions in modern populations have changed).
We use a simulation-based approach to generate population histories accounting for genetic drift, mutation and recombination using the coalescent simulator msprime [26]. This generates (correlated) genealogies for every genetic loci for each individual of both a reference population and a Bottlenecked population. The population structure is simulated such that the ancestral population splits at some time T in the past, taken to be 200 generations in the base case, when some members form a sub-population that remains isolated from the Reference population, as shown in Fig. 1b. Typically, the size of the Reference population, N0, is large relative to the Bottlenecked population, N, taken to be 10,000 and 1000 respectively (unless otherwise stated).
To generate a phenotype, we choose a random sample of SNPs to be causal, and assume that the frequency of these SNPs has been under selection as a result of their effect on the phenotype. A proportion 1 − π of SNPs are assumed to have no causal effect whilst the proportion π (typically small) that contribute to phenotypic variance are distributed as:
$$\begin_\sim N(0,_^),\\ _^=_(1-_)\right]}^\end$$
(1)
where fj is the frequency of the jth mutation in the sample. Because we are dealing with a simulation in which effect sizes are known, we can use a scaled mutation rate µ = πµ0 as the mutation rate for causal SNPs leading to a random number of causal SNPs L with corresponding genotype values xij for individual i at SNP j, where frequencies fj are for the reference population.
This is used to construct a simulated phenotype \(_=_^_}_+_\) [27]. The relationship between effect size and frequency is determined using the parameterisation in which is equivalent to the model used in LDAK [28]. Selection strength (on the trait) is parameterised by s in Eq. 1, and we report results for negative selection (s = −1) so that SNPs with large effect sizes have been selected to become rare. These effect sizes are transferred from the reference to the bottlenecked population, under the assumption of constant environmental variance. The environmental noise ϵi is chosen to control the heritability in the reference population (see Methods) to h2 = 0.5.
Figure 2a, b shows the distribution of heritability in simulated traits from this model for Bottlenecked Populations of different sizes (from N = 500 to 10,000) with T = 200. With the parameters described, the population contraction typically leads to an decrease in the heritability of complex traits. Several factors influence the extent to which heritability is impacted. A more severe contraction leads to lower levels of heritability on average. This is a result of amplified genetic drift due to fewer founding members of the sub-population, resulting in many SNPs being lost. Conversely, the variance increases with contraction size, as there is a possibility of rare SNPs with large effect being able to reach high prevalence.
Fig. 2
Impact of demographic factors of a bottleneck on heritability. a Distribution of heritability at different sizes of bottleneck over 500 repeated simulations. Heritability distributions of populations with initial sizes N = 1000 and N = 8000 with a bottleneck that occurred at T = 200 generations ago. b Mean heritability decreases as the size of the initial Bottlenecked population N decreases. 30 samples were taken at each bottleneck size and the mean and error bars are plotted. c Heritability is distributed with similar tail but different mean and variance over 3 different Bottleneck times. 500 samples are taken for each bottleneck population with different times, T = 50, 200, 500. d There is a positive relationship between the time in the past that the bottleneck occurred, and the variance in heritability. 95% confidence intervals are shown for the 200 samples taken at each time point using an empirical bootstrap
The timing of the contraction can also impact how heritability is distributed. The mean heritability of the complex trait in the bottlenecked population decreases with age as more SNPs are likely to be lost. However, traits vary more when the contraction was longer ago (from T = 50 to 500, Fig. 2c, d) as the most extreme effect size SNPs can become more prevalent as a consequence of genetic drift. Over these time scales, the variance of heritability increases close to linearly with time in the past (Fig. 2d), so heritability becomes increasingly varied for complex traits with equal genomic architecture. For reference, 200 generations corresponds to a split time of around 6000 years, similar in scale to the origin of the Ashkenazi Jewish population; 500 generations corresponds to 15 kya, around the time of the migration to the Americas; and the out-of-Africa bottleneck took place of the order of 2000 generations (60 kya) ago.
Introducing migration rate, m, from the reference population to the bottlenecked population offsets the impact of the contraction on the heritability of complex traits [29]. There is clear dependence between the time of the contraction and the impact of migration (from m = 0–0.1) on mean heritability of traits (Fig. 3a), with older events being more effected. Only modest migration (m = 0.01 with these parameters) is required to mitigate the effect of the contraction. For some problems even ‘one individual per generation’ [30] is enough migration to offset drift, for complex traits the threshold becomes age-dependent as drift interacts with replenishment of variability.
Fig. 3
Growth, migration and time since bottleneck change heritability. a Mean heritability in the Bottlenecked population as migration rate m is increased. 400 samples are taken at each time point and the mean with error bars is plotted. b Effect of bottleneck size and growth on heritability. Shown are the mean and standard errors based on 400 repeated simulations. The reference population is controlled to have heritability 0.5 throughout
Population growth also impacts heritability change in the population after a contraction. As observed in Fig. 3b, whilst growth following a bottleneck does recover some heritability, this is dominated by the strength of the contraction. It should be noted that the total bottlenecked population size can considerably exceed the reference population size in these figures - the population grows by a factor of nearly 20 in 200 generations with a growth rate of 0.015.
Impacts for GWAS inferenceWe have assumed in the simulation study that the effect size of SNPs for a particular complex trait are known. However, in reality effect sizes are inferred from SNP-trait association data using GWAS, which results in constraints being placed on what is known. It might therefore be feared that the outcomes of this paper cannot be directly applied to current complex traits, which we explored in [14]. There are 2 main issues that impact inference in real studies. The first of these is linkage disequilibrium which causes difficulties in isolating causal SNPs. We do not address this problem in this paper but it is explored elsewhere, for example by fine mapping, e.g. in [31, 32], and is unlikely to prevent the genetic drift we describe here.
The second issue pertains to the lack of statistical power of the data in real GWAS studies, weakening the power to resolve many traits. Whilst the models for which our genetic architecture was introduced [28] already considered missing heritability, these models assume selection acts to create the distribution of effect sizes relating to frequency that are present in the observed population. However, that distribution changes by genetic drift. To explore this further, we considered only SNPs that surpass a threshold of detectability in the bottlenecked population, either by conducting GWAS and ranking by p-value or using variance explained vj = fj(1 − fj) \(_^\) in the trait. We considered only the top SNPs by choosing a variance explained threshold over a large number of simulations. For example, when we report the top 1/25 SNPs, we consider the top ∼ 3% of SNPs when averaged over 200 traits. As shown in Fig. 4, the heritability distribution can be effected by limited detection power. Specifically, for traits with low heritability in the target population, measured heritability for thresholded SNPs drops, as there are few (or even no) remaining SNPs that have sufficient explanatory power to be recorded. However, for traits with high observed heritability, the true heritability remains high. It follows that our observations on the impact of genetic drift on selection are valid for traits with high heritability, and increasingly for traits with lower heritability as GWAS sample size grows. This also implies that (as is widely assumed) there will be a number of traits that are highly heritable, but that are more difficult to study in drifted (e.g. European) populations.
Fig. 4
The distribution of heritability of a bottlenecked population as observable in a GWAS study against simulated heritability present, over 200 repeated experiments. a the simulated heritability distribution (yellow) and observed thresholded heritability (green) where only the 1/29 percentile of most informative SNPs (by VE = variance explained) are observed. b Comparing heritability when thresholding on p-values over 200 repeated experiments, where the threshold heritability only captures the t = 1/2n most extreme SNPs. The red line depicts values such that simulated and threshold heritability are equal. c as b, but where SNPs are ranked by variance explained. d Pearson correlation and regression coefficient between the real heritability and threshold heritability for thresholds t = 1/2n with standard error bars
留言 (0)