Cell lines, culture media, and reagents for treatment
HeLa (C01-CA) human cervical adenocarcinoma cells and SiHa (C01-FH) human squamous carcinoma cells were obtained from Novobio (Shanghai, China) and cultured in DMEM and RPMI-1640 medium containing 10% fetal bovine serum (FBS, EVERY GREEN, Zhejiang, China), respectively. The cells were then incubated in a humidified atmosphere at 37 °C with 5% CO2. Selenium dioxide (Aldrich-200107) and GSK-J4 Hcl (S7070, a specific inhibitor of JMJD3/UTX), were purchased from Selleck (Houston, USA).
Cell viability assay
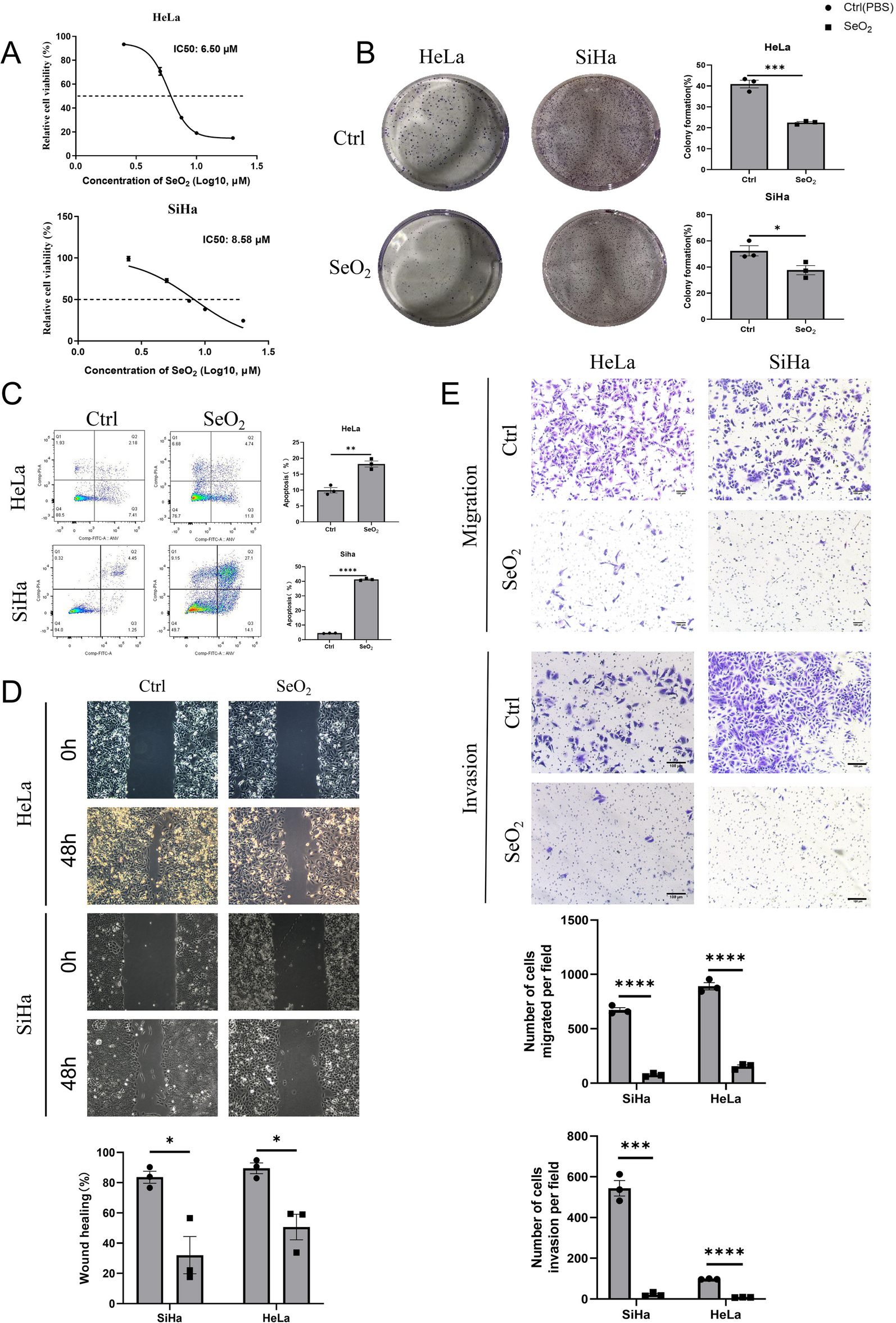
The pharmacological effects of SeO2 and GSK-J4 on cell proliferation were assessed using Cell Counting Kit-8 (CCK-8, TransGen, Beijing, China) following the manufacturer's instructions. Briefly, HeLa and SiHa cells were seeded at a density of 3 × 103 cells/well in 96-well culture plates and treated with varying concentrations of SeO2 and GSK-J4. After treatment, 20 μl of CCK-8 solution was added to each well and incubated for an additional 2 h at 37 °C in a 5% CO2 incubator. The optical density (OD) of each well was measured at 450 nm using a microplate reader (BioTek, VT, USA), and the data was graphically displayed. The growth rate (%) was calculated using the formula: (ODExperiment − ODBlank)/(ODControl − ODBlank) × 100%. The IC50 was determined using GraphPad Prism 9 software (GraphPad Software, USA).
Colony formation assay
Cells were seeded in 6-well plates at a cell density of 1000 cells/mL or 500 cells/mL, respectively. After 48 h of culture in normal medium, the culture medium was replaced with SeO2/GSK-J4 or a solvent control of PBS/DMSO, and the medium was changed every 3 days. After 7 days, the cells were washed with PBS, fixed with 4% paraformaldehyde, and stained with 0.1% crystal violet solution. Colonies were counted using ImageJ software [23].
Flow cytometry assay
To assess the cell cycle distribution and cellular apoptosis, 7 × 104 cells were seeded into a 6-well plate and treated with SeO2 and GSK-J4. The samples for the cell cycle analysis were stained with propidium iodide (BD Biosciences, MA, USA), and the samples for the apoptosis analysis were stained with Annexin V-fluorescein isothiocyanate and propidium iodide (TransGen, Beijing, China). Flow cytometric analysis was performed using a BD FACSCanto II™ flow cytometer (BD Biosciences, MA, USA). The cell cycle distribution was analyzed with ModFit LT V3.1 (Verity Software House, USA) software and the apoptotic rate of cells was evaluated with FlowJo V10.0.7 (Treestar Inc., Ashland, OR, USA) software.
Scratch wound healing assay
Cells were seeded onto 24-well plates and treated with SeO2 and GSK-J4 or a solvent control of PBS/DMSO. Prior to treatment, a clean line was created using a sterile 200 μl pipette tip. The migration of cells was observed under an inverted microscope (Leica DMI6000B, GER) and imaged at 0 h and 48 h. The relative scratch area was calculated using ImageJ software. Wound healing percentage (%) was calculated using the formula: 1 − (S48h/S0h) × 100%, where S0h represents the initial scratch area at 0 h, and S48 h represents the scratch area at 48 h.
Transwell chamber assay
Transwell chambers (Corning, USA) with polycarbonate membranes (8.0 μm pore size) were inserted into a 24-well culture plate. For the migration assay, HeLa cells (5 × 104/well) and SiHa cells (3 × 104/well) were plated into the upper chambers with 200 μl FBS-free medium containing SeO2 and GSK-J4 or a solvent control of PBS/DMSO. For the invasion assay, the upper chamber was evenly coated with 100 μl of Matrigel (Corning, USA) and an FBS-free medium mixture (1:8), and the remaining steps were analogous to those of the cell migration tests. Next, 800 μl of complete medium (10% FBS) was added to the lower chamber as a chemoattractant. After incubation at 37 °C in 5% CO2, the cells on the upper surface of the filters were removed with a cotton swab, and the invading cells on the lower surface were fixed with 4% paraformaldehyde for 30 min and stained with a 0.1% crystal violet solution for 30 min. The migration and invasion cells on the lower surface of the membrane in each chamber were counted randomly under high power fields.
RNA extraction, reverse transcription, and quantitative real-time PCR
After treatment with either solvent control PBS/DMSO or SeO2/GSK-J4 for different time periods, total RNA was extracted from cells using RNAiso Plus Reagent (Takara, Japan) in accordance with the manufacturer's instructions. Next, 1 μg of total RNA was reverse transcribed into cDNA using a reverse transcriptase Kit (TransGen, Beijing, China). The mRNA expression was determined using real-time PCR with the PerfectStartTM Green qPCR SuperMix Kit (TransGen, Beijing, China) on a StepOne Plus PCR system (Applied Biosystems, Waltham, MA, USA). The 2−ΔΔCT formula was used to calculate the expression of the genes, with the human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene serving as the internal reference. The primer information is listed in Additional file 1: Table S1.
Western blotting
Whole-cell lysates were obtained from cells treated with PBS/DMSO or SeO2/GSK-J4 using RIPA lysis buffer supplemented with a protease inhibitor cocktail. The lysates were then sonicated and centrifuged, and the supernatant was mixed with loading buffer and heated. Equal amounts of protein samples were separated by SDS-PAGE and transferred to PVDF membranes. After blocking with 5% skim milk, the membranes were incubated with primary antibodies overnight at 4 °C, followed by secondary antibodies and detection using an enhanced chemiluminescence (ECL) kit. Antibodies against KDM6B/JMJD3, H3K27me1, Bax, Bcl-2, Caspase-7, KDM6A/UTX, H3K27me3, Cyclin B1, Caspase-3, E-cadherin, N-cadherin, Vimentin, GAPDH, β-Tubulin, MMP-1, MMP-2, MMP-9, and CDK1 were used. The blot signals were detected using an Ultrasensitive ECL Detection Kit (Proteintech, Wuhan, China) and visualized by the ImageQuant LAS 500 (Cytiva, USA).
The antibodies used were: KDM6B/JMJD3 (1:1000, ab169197), H3K27me1 (1:500, ab194688), Bax (1:500, ab53154), Bcl-2 (1:1000, ab196495), and Caspase-7 (1:200, ab25900) from Abcam (Cambridge, UK); KDM6A/UTX (1:500, PA5-31828) from Invitrogen-Thermo Fisher Scientific (CA, USA); H3 (1:2000, #4499), H3K27me3 (1:1000, #9733), Cyclin B1 (1:1000, #12231), Caspase-3 (1:1000, #9662S), E-cadherin (1:1000, #3195), N-cadherin (1:1000, #13116), Vimentin (1:1000, #5741), GAPDH (1:1000, #2118), and β-Tubulin (1:1000, #2128) from Cell Signaling Technology (MA, USA); and MMP-1 (1:500, #10371-2-AP), MMP-2 (1:500, #10373-2-AP), MMP-9 (1:500, #10375-2-AP), CDK1 (1:500, #19532-1-AP), and horse radish peroxidase (HRP)-conjugated goat secondary antibodies against rabbit and mouse from Proteintech (Wuhan, China).
Immunofluorescence staining
Cells were plated on glass coverslips in 24-well culture plates and treated with SeO2 and GSK-J4 or PBS/DMSO. After removing the cell supernatant, the cells were washed twice with cold PBS and fixed in 4% paraformaldehyde at 4 °C for 15 min. They were then permeabilized with 0.5% Triton X-100 at room temperature for 10 min and blocked with 2% BSA at 37 °C for 30 min. The coverslips were incubated with primary antibodies against Ki67 (1:400, Cell Signaling Technology, #9129, USA) and H3K27me3 (1:800, Cell Signaling Technology, #9733S, USA) overnight at 4 °C. After incubation with Alexa Fluor 488-conjugated secondary antibody (1:500, Abcam, ab150077, UK) at 37 °C for 1 h in the dark, the nuclei were stained with DAPI for 20 min at room temperature and washed with PBS. Staining was observed with a Confocal Laser Scanning Microscopy (CLSM, FV3000, Olympus, Tokyo, Japan), and the fluorescence intensity was analyzed by OLYMPUS cellSens 1.16 software.
Chromatin immunoprecipitation (ChIP) assay
For the ChIP assay, the Simple ChIP Plus Enzymatic Chromatin IP Kit (Cell Signaling Technology, #9005, USA) was used according to the manufacturer's instructions. Cells were cross-linked using formaldehyde and then incubated with glycine. Chromatin was digested with Micrococcal Nuclease, and the nuclei were lysed using a VCX130 Sonicator. The chromatin-protein complexes were immunoprecipitated using anti-H3K27me3 (1:50, Cell Signaling Technology, #9733, USA) antibody, with normal rabbit IgG antibody (Cell Signaling Technology, #2729, USA) serving as the negative control. Purified DNA fragments were used as templates for RT-qPCR with primers listed in Additional file 1: Table S2.
The generation, identification, and treatment of human cervical cancer-derived organoids (HCCOs)
HCCOs were generated using tissue samples obtained from the Obstetrics and Gynecology ward at Ningbo Medical Center, Lihuili Hospital. These samples were collected from surgical patients who had not undergone preoperative radiotherapy or chemotherapy. The tissue specimens were divided into three parts: one for paraffin embedding, one for cryopreservation in liquid nitrogen, and one for organoid construction. The tumor tissue was minced and digested with collagenase for 30 min. The resulting tissue clusters were then filtered through a 100 μm filter and embedded in a matrix called Matrigel (Corning, 356231, USA). Each well containing the embedded tissue clusters was supplemented with cervical cancer organoid medium, which was changed every 3–4 days. The medium used for culturing the HCCO was obtained from Absin (Shanghai, abs9590, China). The HCCO culture medium composition included DMEM/F12 supplemented with HEPES, N2, B27, EGF, Wnt3a, Primocin, Glutamax, Gentamicin/amphotericin B, Noggin, R-spondin 1, E2, Recombinant human A83-01, Y-27632 HCL, human FGF-10, and human FGF-β. After seeding the organoids in 24-well culture plates and amplifying them, HCCOs and the original tissues were embedded in paraffin and subjected to Hematoxylin and Eosin (HE) staining. Following that, Short Tandem Repeat (STR) DNA sequences were performed on both patient tumors and organoids to confirm the identities of the organoids. Subsequently, the HCCOs were treated with selenium or GSK-J4 at the indicated concentrations, and the growth status of the organoids was monitored. On the third day, the activity of the HCCOs derived from squamous cell carcinoma was assessed using the CCK-8 reagent, following the same methodology employed in the cell experiments. The activity of HCCOs from adenocarcinoma was assessed through conducting viable and non-viable cells staining using a Calcein AM/PI Cell Live/Dead Assay Kit (Beyotime, C2015S, China) according to the instruction from the manufacture.
Establishment of three-dimensional bio-printed cervical cancer models and in vitro culture
According to Xie et al. [24], specimens were well minced and washed twice with DMEM/F12 (GIBCO) plus 1% penicillin/streptomycin (Sigma), 1% GlutaMAX (GIBCO), and 10-mM HEPES (GIBCO). The minced tissue was then digested into a single-cell suspension by incubating with EBSS (HyClone) containing 2.5 mg/ml collagenase D, (Roche) and 0.1 mg/ml DNaseI(Sigma) for 2–6 h and the digestion was stopped by adding cold advanced DMEM/F12. The cell suspension was then filtered through a 70 μm nylon cell strainer, and the yield cells were collected and resuspended into the culture medium into a concentration of 6.0 × 106/ml.
To formulate the bioink for printing, the cell suspension was mixed up with buffer solution containing 5% gelatin (Sigma) and 1% sodium alginate (Sigma), resulting in a final cell concentration of 3.0 × 106/ml. The bioink was drawn into a 1 ml syringe with a 23-gauge needle and printed using the 3D Cell Printer by SUNP Co. The models were printed in layers, with each model being a cube measuring 10 mm × 10 mm × 1.2 mm and consisting of four layers of bioink. The bioink contained a precise cell number of 2.5 × 105. The shape of the HCCO models was designed as a square grid with independent holes between the frames to maximize the contact area between the cells and the culture medium. The models were collected using twelve-well plates and were used for viability tests. After printing, the HCCO models were fixed with a calcium chloride (CaCl2) solution for 1 min to provide better strength. Then, the models were supplemented with 2–3 ml of the HCCO culture medium. The culture medium was changed three times a week, and each time the models were fixed with calcium chloride.
Subcutaneous xenograft model
Female BALB/C nude mice were obtained from Hunan Silaikejingda Laboratories (Hunan, China) for the xenograft mouse model. The mice were allowed to acclimatize to the laboratory environment for 1 week prior to the experiments and provided with ad libitum access to food and water. To establish BALB/c nude mice bearing tumors, SiHa cells (5 × 106) were inoculated into the right flank of the mice in 200 μl volumes per site. On the seventh day after the injection of tumor cells, the mice were randomly divided into two groups: the sham-treated group and the SeO2/GSK-J4 treated group. The mice in the SeO2/GSK-J4 treated group received intraperitoneal injections of SeO2 (3 mg/kg) or GSK-J4 (50 mg/kg), while the sham-treated group received an equal volume of PBS/DMSO. These injections were administered every 2 days. Tumor length (L) and width (W) were measured using calipers every 2 days, and tumor volumes were calculated using the equation (L × W2)/2. After 18 days of treatment, the mice were euthanized humanely by cervical dislocation, and all tumors were collected for further analysis.
Paraffin-embedded cervical tissue specimens and Immunohistochemical stain
We gathered paraffin-embedded cervical tissue specimens from the Department of Pathology archives of the First Affiliated Hospital of Nanchang University for JMJD3/UTX immunohistochemistry assays (IHC). The specimens included normal cervical tissues (n = 4), cervical squamous cell carcinoma (SqCa, n = 14), and endocervical adenocarcinoma (AdCa, n = 4). The pathology team at the hospital identified the tumor and normal tissues from the hysterectomy samples. Carcinoma samples were obtained from patients who were classified as stage I B to II A according to the 2009 International Federation of Gynecology and Obstetrics (FIGO) classification. None of the patients had undergone neoadjuvant chemotherapy or radiotherapy prior to radical operation.
Immunohistochemistry was utilized to evaluate the expression of KDM6A and KDM6B proteins in paraffin-embedded cervical tissue samples. The samples were deparaffinized and rehydrated, followed by antigen retrieval. Slides were then incubated with primary antibodies against UTX/KDM6A (1:500) and JMJD3/KDM6B (1:200), followed by secondary antibodies and staining with DAB. Positive staining appeared as brown staining in the cell membrane or nucleus. Image quantification was performed using ImageJ software.
Statistical analysis
Data are presented as the mean ± standard Error (SEM), and all graphs and statistical analyses were conducted with GraphPad Prism 9. Statistical difference between two groups were compared using unpaired/paired Student’s t-test or one-way ANOVA. A value of p < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).

留言 (0)