
記住我




Malignant hyperthermia, a life-threatening pharmacogenetic condition triggered by volatile anaesthetics and/or succinylcholine, occurs in 1 : 10 000 to 1 : 250 000 procedures, with half of all cases reported in children and young people.1,2 Onset of malignant hyperthermia can be sudden and progression rapid, with potentially fatal outcomes.1 Dantrolene sodium is an effective emergency treatment for malignant hyperthermia; however, the risk of complications, their severity and the length of stay in intensive care increase with every 15 to 30 min delay between the first signs of malignant hyperthermia and dantrolene administration.3–5 European Malignant Hyperthermia Group (EMHG) guidelines recommend rapidly administering 2 to 2.5 mg kg−1 intravenous (i.v.) dantrolene upon recognising the condition, with repeat dosing every ∼10 min until the reaction is controlled, noting that doses of greater than 10 mg kg−1 may be required.6 These guidelines advise precautionary dantrolene stocking based on the DANTRIUM® IV (also marketed as DANTROLEN IV; Norgine, Harefield, UK) formulation available in Europe.6 Dantrium® is supplied in vials containing 20 mg dantrolene for reconstitution in 60 ml water by vigorous shaking; because of the poor solubility of dantrolene, preparation of a single vial can take more than 4 min.7,8 For a 60 kg patient, effective doses may require 8 to greater than 30 vials, requiring the effort of at least three clinical staff and potentially contributing to critical delays.6–8 To shorten the preparation and administration time and facilitate rapid interventions in emergency situations, a novel intravenous formulation of dantrolene, NPJ5008, has been developed using different excipients [2-hydroxypropyl-beta-cyclodextrin (HP-β-CD) and polyethylene glycol (PEG)]. With NPJ5008, the same dose of dantrolene can be reconstituted more rapidly and in smaller volumes than with Dantrium® (unpublished observations). Standard preclinical assessment of NPJ5008 in rats revealed no significant differences to Dantrium® (unpublished observations).
We report a phase 1 clinical safety trial in healthy adult volunteers, evaluating the pharmacokinetics, including bioequivalence, of NPJ5008 versus Dantrium®, and assessing the safety and tolerability profile of NPJ5008. To evaluate the time-saving benefits of the novel formulation, trial pharmacy product preparation data were analysed; however, as the measured conduct of the volunteer trial necessarily differed from the emergency administration of dantrolene in malignant hyperthermia, only timings for discrete processes were considered representative. A laboratory simulation focusing on speed, thus more reflective of a malignant hyperthermia emergency situation, directly compared preparation and administration times for the two formulations.
The Consolidated Standards of Reporting Trials (CONSORT) guidelines were followed in this study: Schulz KF, Altman DG, Moher D, for the CONSORT Group. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials.
Methods EthicsThis single-centre study received approval with the protocol number NPJ5008-01/2020 from The Office for Research Ethics Committees Northern Ireland, Lisburn BT28 2RF (Chairman Dr Alastair Walker) on 2 February 2021. A substantial protocol amendment was approved (before the commencement of the study) by ORECNI Chairman Barry Mimnagh. Authorisation from the UK's Medicines and Healthcare products Regulatory Agency was received on 12 March 2021. The study followed relevant regulatory requirements and the ethical standards of Directive 2001/20/EC (www.ema.europa.eu/en/ich-e6-r2-good-clinical-practice-scientific-guideline; www.legislation.gov.uk/uksi/2004/1031/contents/made; www.legislation.gov.uk/uksi/2006/2984/contents/made; www.legislation.gov.uk/uksi/2006/1928/contents/made; www.legislation.gov.uk/uksi/2008/941/pdfs/uksi_20080941_en.pdf; www.legislation.gov.uk/uksi/2019/744/made). The trial took place at Quotient Sciences (Ruddington, UK). All participants provided written informed consent in accordance with the Declaration of Helsinki (www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects).
Study populationHealthy adult volunteers aged 18 to 55, weighing at least 55 kg, with a body mass index (BMI) between 19.0 and 32.0 kg (m2)−1 were eligible provided they passed preliminary screening. Only women of nonchildbearing potential were enrolled (as data on dantrolene use in pregnant women are limited) and male individuals were required to adhere to contraception directives (www.medicines.org.uk/emc/product/1097/smpc#gref).9 Key exclusion criteria included: exposure to any investigational medicinal product within defined time frames; drug abuse; alcohol abuse; smoking; history or symptoms of significant medical conditions, including estimated glomerular filtration rate of less than 30 ml min−1 (1.73 m2)−1 or impairments in respiratory/airway function; abnormal blood parameters; history of adverse reaction to any drug/allergen and taking any prohibited concomitant medication/herbal remedies within specified periods.
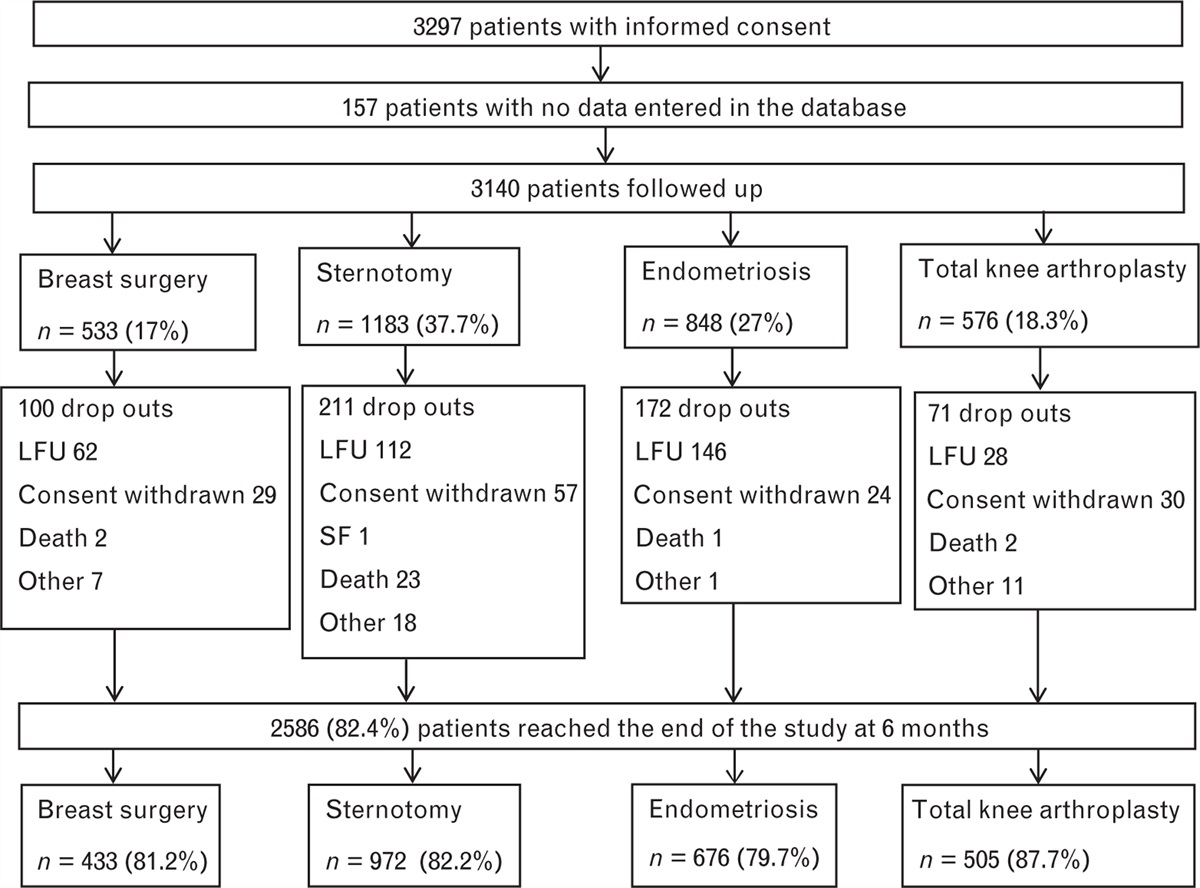
Study design, objectives and endpointsThe study comprised two open-label, single-dose parts (see Fig. 1 for participant flow). Doses of 60 and 120 mg NPJ5008 were selected equating to 1 and 2 mg kg−1, respectively, for a 60 kg person (rounded average adult weight worldwide),10 i.e. the initial dose of dantrolene recommended in some countries (e.g. Japan) and the lower end of EMHG dose recommendation, respectively.6,11 These doses were set below the maximum tolerability threshold for healthy volunteers reported previously (www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205579Orig1s000SumR.pdf).
 Fig. 1:
Fig. 1: Part 1 and Part 2 participant flow diagram.
Part 1 was a two-period crossover study with participants randomised 1 : 1 to the two treatment sequences using computer-generated randomisation. The primary objective was to assess the bioequivalence, in terms of overall exposure, of the NPJ5008 (test) formulation (administered as 11.3 ml of a 5.3 mg ml−1 solution over at least 1 min) versus Dantrium® (the reference product; administered as 186 ml of a 0.32 mg ml−1 solution over at least 5 min), both yielding a dose of 60 mg dantrolene, following standard methods,12 using an Alaris CC infusion pump (Becton Dickinson, Wokingham, UK). The rate of administration of Dantrium® in a clinical trial setting is limited by the speed at which the volume can be delivered by available infusion pumps. In a previous study investigating higher concentration formulations of i.v. dantrolene in healthy volunteers, severe adverse events related to the mode of action (MoA) of dantrolene occurred when 1.75 mg kg−1 was administered over 30 s. No severe adverse events occurred when slower infusion times were used with doses up to 2.5 mg kg−1 (www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205579Orig1s000SumR.pdf). Therefore, infusions in this study were to have a minimum duration of 1 min.
The primary endpoints included area under the concentration–time curve (AUC) from time zero to last measurable concentration AUC0 to last and extrapolated to infinity AUC0 to ∞, but excluded the maximum observed drug concentration in plasma (Cmax), because Cmax values were expected to vary by product infusion rate (60 mg min−1 for NPJ5008 and 12 mg min−1 for Dantrium®).
Secondary objectives of Part 1 assessed the i.v. pharmacokinetics and the safety and tolerability profile of NPJ5008. Secondary plasma pharmacokinetic endpoints included: Cmax; truncated AUCs (time zero to 6 and 72 h post dose); time prior to the first measurable concentration (Tlag); time from drug administration to maximum observed analyte concentration (Tmax); terminal rate constant (λz); terminal half-life of the analyte (T1/2); total body clearance calculated after a single i.v. administration (dantrolene only); and volume of distribution based on the terminal phase calculated using AUC0 to ∞ after a single administration (Vz, dantrolene only), including statistical assessment of relative bioavailability.
Part 2 started at least 72 h post dose of Part 1, following a data review, and assessed the safety and tolerability profile of a 120 mg dose of NPJ5008 administered as 22.6 ml of a 5.3 mg ml−1 solution (Alaris CC infusion pump). The relative bioavailability of 60 versus 120 mg NPJ5008 was an exploratory objective.
Pharmacokinetic parameters were estimated at selected times before dosing and at 20 time points until 72 h post dose by noncompartmental analysis using Phoenix WinNonlin software (v8.0 and v8.3, Certara USA, Inc., Princeton, New Jersey, USA).
Bioanalytical methodsThe plasma concentrations of dantrolene (free acid) and 5-hydroxydantrolene (a pharmacologically active metabolite) were determined using a validated analytical method using liquid chromatography with tandem mass spectrometry detection at Labcorp Drug Development (Norgine Pharmaceuticals Ltd data on file). The lower limit of quantification was 10 ng ml−1 for dantrolene and 1 ng ml−1 for 5-hydroxydantrolene.
Statistical methodsThe sample sizes were based on regulatory guidance and the possibility of drop-out for Part 1, and on previous experience with similar studies for Part 2. The sample size for Part 1 exceeded that calculated to have at least 80% power to conclude equivalence based on two one-sided t tests with a type I error rate of 0.05 [resulting in 90% confidence intervals (CIs)], assuming a ratio of means of 1.05, intra-individual variability of 11% and standard acceptance limits for equivalence. The assumed variability was based on data for Dantrium® (www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205579Orig1s000ClinPharmR.pdf).
The pharmacokinetic parameters were log-transformed prior to analysis. Statistical analyses utilised analysis of variance with mixed-effects modelling, which included terms for period, sequence and regimen as fixed effects and individual within sequence as a random effect. The ratios of the geometric means (GMR) of test versus reference and their two-sided 90% CIs were calculated for all primary and secondary pharmacokinetic endpoints. Bioequivalence was evaluated using the average bioequivalence method: if the 90% CI for the GMRs for each of the primary endpoints fell within the prespecified standard 80 to 125% acceptance range on formal statistical analysis, then the null hypothesis of nonequivalence was rejected.12 This analysis strategy follows relevant guidelines (www.fda.gov/regulatory-information/search-fda-guidance-documents/statistical-approaches-establishing-bioequivalence; www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf). Assessment of relative bioavailability was performed on secondary pharmacokinetic parameters with no correction for nominal dose received (D) using the same model as for bioequivalence (Part 1 only) and corrected for D between the 60 and 120 mg NPJ5008 dose levels, analysed as above, in a model that included terms for regimen as a fixed effect and body weight as a covariate. Pharmacokinetic parameters were analysed descriptively, with geometric coefficients of intra-individual variability (CVw) measuring dispersion. The model was fitted using SAS Software PROC MIXED (version 9.4, SAS Ltd, Marlow, UK) specifying the restricted maximum likelihood method.
More details are available in the Supplemental Digital Content, https://links.lww.com/EJA/A923.
Safety assessmentSafety evaluations comprised analysis of adverse events, laboratory variables (haematology, clinical chemistry and urinalysis) and vital signs monitoring at specified intervals, including continuous peripheral oxygen saturation (pulse oximeter), 12-lead ECGs, hand grip tests (Jamar Hand Dynamometer, Williams Medical Supplies, Rhymney, UK), physical examinations and spirometry, using published methods to calculate and adjust predicted values.13 Adverse events were coded and classified according to seriousness, severity and relationship to the study drug, determined primarily in terms of the temporal relationship with dosing and the known MoA of the drug (www.ema.europa.eu/en/ich-e2a-clinical-safety-data-management-definitions-standards-expedited-reporting-scientific; www.meddra.org/news-and-events/news/english-meddra-version-240-now-available-download).
Pharmacy report analysisData were gathered on discrete ‘start’ and ‘end’ time points within the preparation and administration process of NPJ5008 and Dantrium® from trial pharmacy records. Only interval timings with an identical and discrete process for all three regimens were analysed.
Laboratory simulation of preparation and administrationFive vials each of the two dantrolene formulations were reconstituted and delivered as swiftly as possible via suitable syringes, and either a 16-gauge or 22-gauge cannula, to simulate an i.v. push in adults and children, respectively. Infusion pumps were not used. Data were gathered on preparation [of water for injection (WFI), reconstitution, draw up] and administration times. Additional study details are available in the Supplemental Digital Content, https://links.lww.com/EJA/A923.
Results ParticipantsEnrolment started on 2 April 2021 and the study continued until July 2021 (see Fig. 1 for participant flow).
For Part 1, 16 healthy volunteers (one female) were enrolled, dosed per the assigned treatment sequence and completed the study. All were non-Hispanic white, aged 23 to 55 years and weighed 61.5 to 105.5 kg (median 82.05 kg), with a BMI of 20.4 to 29.5 (median 25.25). The mean age was higher in the treatment sequence test-reference than in the reverse sequence (44.3 vs. 34.6 years), but there were no other notable group differences.
For Part 2, five participants (one female) aged 20 to 55 years who enrolled completed the study. All were non-Hispanic white, weighed 83.2 to 92.4 kg (median 85.8 kg), with a BMI of 25.1 to 29.4 (median 27.90).
Bioequivalence results (primary objective)On average, overall dantrolene (free acid) exposure following 60 mg NPJ5008 administration was 90.24 and 90.44% of that following administration of 60 mg Dantrium®, as measured by AUC0 to last and AUC0 to ∞, respectively (see Table 1). Formal statistical analysis indicated 90% CIs of the ratios were within the prespecified 80 to 125% bioequivalence limits, demonstrating bioequivalence of NPJ5008 and Dantrium® in terms of overall exposure.
Table 1 - Results of bioequivalence assessment for dantrolene (free acid) following single intravenous infusion of 60 mg Dantrium® or 60 mg NPJ5008 in healthy volunteers (n = 16) Parameter Adjusteda geometric mean NPJ5008 (test) (n = 16) Adjusteda geometric mean Dantrium® (reference) (n = 16) Ratio (test : reference) 90% CI Largest P value from two one-sided tests CVw AUC0 to last (ng h ml−1) 11 900 13 200 90.24% 85.94 to 94.76% <0.001 7.85% AUC0 to ∞ (ng h ml−1) 12 300 13 600 90.44% 85.97 to 95.14% <0.001 8.16%AUC0 to ∞, area under the curve from time zero extrapolated to infinity; AUC0 to last, area under the curve from time zero to last measurable concentration; CI, confidence interval; CVw, geometric coefficient of intra-individual variability.
aThe model was adjusted for fixed effects of period, regimen and sequence, and individual within sequence as a random effect. Of note there was a significant period effect, P = 0.021 for AUC0 to last and P = 0.044 for AUC0 to ∞, but no significant sequence effect was seen, therefore, the balanced crossover design meant period had no material impact on the results.
The mean plasma concentration–time profiles for NPJ5008 and Dantrium® were similar (Fig. 2a), and had comparable Tmax ranges, Cmax and resultant T1/2 (Table 2).
 Fig. 2:
Fig. 2: Mean plasma concentration–time profiles on log10/linear scale for dantrolene (free acid) and the 5-hydroxydantrolene metabolite following single intravenous infusion doses of (a) 60 mg NPJ5008 or 60 mg Dantrium® and (b) 60 mg NPJ5008 or 120 mg NPJ5008.
Table 2 - Plasma pharmacokinetic parameters for dantrolene (free acid) and 5-hydroxydantrolene following single intravenous infusion of 60 mg Dantrium®, 60 mg NPJ5008 and 120 mg NPJ5008 in healthy volunteers Analyte Dantrolene 5-Hydroxydantrolene Treatment dose level Dantrium® 60 mgan = 16 NPJ5008 60 mgan = 16 NPJ5008 120 mgbn = 5 Dantrium® 60 mgan = 16 NPJ5008 60 mgan = 16 NPJ5008 120 mgbn = 5 T max (h) 0.155 [0.14 to 4.16] 0.100 [0.01 to 4.01] 0.149 [0.07 to 0.80] 10.135 [1.64 to 16.15] 6.017 [4.00 to 10.02] 10.064 [10.05 to 16.17] C max (ng ml−1) 1170 (34.8) 1090 (29.7) 2090 (24.2) 138 (34.1) 114 (25.3) 208 (22.5) C max/D c (ng ml−1 mg−1) 24.9 (34.8) 23.1 (29.7) 22.1 (24.2) 2.93 (34.1) 2.40 (25.3) 2.20 (22.5) AUC0 to 6 (ng h ml−1) 4790 (18.8) 4230 (15.4) 9400 (25.7) 581 (40.0) 497 (27.4) 765 (17.4) AUC0 to 72 (ng h ml−1) 13 500 (23.9) 12 200 (20.8) 30 500 (31.1) 3380 (21.2) 2890 (19.9) 6150 (23.6) AUC0 to last (ng h ml−1) 13 200 (23.9) 11 900 (20.6) 30 400 (31.7) 3370 (21.1) 2880 (19.6) 6150 (23.6) AUC0 to last/Dc (ng h ml−1 mg−1) 279 (23.9) 252 (20.6) 322 (31.7) 71.4 (21.1) 60.9 (19.6) 65.2 (23.6) AUC0 to ∞ (ng h ml−1) 13 600 (24.2) 12 300 (21.1) 30 900 (31.5) 3420 (20.8) 2920 (19.9) 6270 (23.9) AUC0 to ∞/Dc (ng h ml−1 mg−1) 287 (24.2) 260 (21.1) 327 (31.5) 72.4 (20.8) 61.9 (19.9) 66.4 (23.9) T 1/2 (h) 8.476 (21.8) 9.041 (27.9) 10.993 (18.5) 9.488 (24.0) 10.171 (20.3) 11.212 (19.4) λz (h−1) 0.08178 (21.8) 0.07667 (27.9) 0.06305 (18.5) 0.07306 (24.0) 0.06815 (20.3) 0.06182 (19.4) CL (ml min−1) 58.2 (24.2) 63.5 (21.1) 50.5 (31.5) NC NC NC Vz (l) 42.7 (21.1) 49.7 (22.7) 48.0 (29.2) NC NC NC Vss (l) 46.3 (16.6) 53.3 (14.9) 49.2 (24.5) NC NC NC MPR C max (ratio) NC NC NC 0.112 (37.4) 0.099 (32.8) 0.095 (35.6) MPR AUC0 to last (ratio) NC NC NC 0.243 (30.3) 0.230 (23.3) 0.193 (24.0) MPR AUC0 to ∞ (ratio) NC NC NC 0.240 (29.9) 0.227 (22.9) 0.193 (23.7)Data are median [range] and geomteric mean (geometric coefficient of variation%).λz, first order rate constant associated with the terminal (log-linear) portion of the curve; AUC, area under the curve; AUC0 to ∞, area under the curve from time zero extrapolated to infinity; AUC0 to last, area under the curve from time zero to last measurable concentration; CL, total body clearance calculated after a single i.v. administration; Cmax, maximum observed drug concentration in plasma; D, dose; i.v., intravenous; MPR, metabolite to parent ratio; NC, not calculated; T1/2, terminal half-life of the analyte in plasma; Tmax, time from administration to maximum observed concentration of the analyte in plasma; truncated AUCs, time zero to 6 and 72 h post dose; Vss, predicted volume of distribution at steady state after single i.v. administration; Vz, volume of distribution based on the terminal phase calculated using AUC0 to ∞ after a single i.v. administration.
aIn Part 1, the actual dose received (based on the products’ Certificate of Analysis, which differed slightly from the nominal dose, and participant weight) ranged from 0.56 to 0.97 mg kg−1 of NPJ5008 and from 0.57 to 0.98 mg kg−1 of Dantrium®.
bIn Part 2, a range of actual doses of 1.29 to 1.43 mg kg−1 of NPJ5008 were administered. Data presented are median [range]; all other data presented are geometric mean (geometric CV%).
cDose (D)-corrected parameters were normalised by the nominal salt-corrected dose, for example, 120 mg × 0.787 (salt correction factor) = 94.44 mg.
The relative bioavailability for NPJ5008 of dantrolene (free acid) Cmax was comparable to Dantrium®, with an adjusted GMR of 92.76% (90% CI 78.27 to 109.93%, P = 0.45). In terms of truncated AUC assessments, the relative bioavailability of NPJ5008 was significantly lower than for Dantrium®, with adjusted GMRs of 88.35% (90% CI 84.95 to 91.89%, P < 0.001) and 90.27% (90% CI 85.89 to 94.88%, P = 0.003) for AUC0 to 6 and AUC0 to 72, respectively.
The mean plasma concentration–time profiles and resultant pharmacokinetic parameters of the major metabolite 5-hydroxydantrolene were similar for NPJ5008 and Dantrium®, with similar elimination times (T1/2) but different peak times (Tmax) (Fig. 2a, Table 2). The geometric metabolite to parent ratios (MPRs) were similar following administration of NPJ5008 and Dantrium®: 0.099 and 0.112 based on Cmax and 0.230 and 0.243 based on AUC0 to ∞, respectively. Overall exposure of 5-hydroxydantrolene as measured across all AUC or Cmax parameters was significantly lower (P values between <0.001 and 0.006) for NPJ5008 compared with Dantrium® (statistical data in Supplemental Digital Content, https://links.lww.com/EJA/A923).
In Part 2, the mean dantrolene (free acid) and metabolite plasma concentration–time profiles and resultant pharmacokinetic parameters for 60 and 120 mg NPJ5008 (Fig. 2b and Table 2) indicate elimination of each analyte was similar at the higher dose (T1/2 ∼11 h), and at both doses, dantrolene exposure was higher than metabolite exposure (MPRs of ∼0.10 for Cmax).
Exploratory assessment of the relative bioavailability of 60 and 120 mg NPJ5008 suggested it was comparable in terms of salt-corrected and dose-corrected Cmax, with an adjusted GMR of 99.26% (90% CI 77.51 to 127.11%, P = 0.96), but was significantly lower for 60 mg than 120 mg for dose-corrected AUC parameters, with adjusted GMRs of 75.21% (90% CI 61.40 to 92.13%, P = 0.026) and 76.15% (90% CI 62.06 to 93.45%, P = 0.033) for AUC0 to last and AUC0 to ∞, respectively.
Safety and tolerability of NPJ5008 at low and high doses (primary/secondary objectives)No deaths or serious adverse events were reported and no participant withdrawal or study stopping criteria were met. The incidence and severity of the adverse events were comparable between 60 mg NPJ5008 and Dantrium®, and in Part 2 were consistent with an increased dose of NPJ5008 (Table 3). All but one participant experienced adverse events (TEAEs), consistent with the MoA of dantrolene. Overall, only one participant (in Part 2) experienced a severe TEAE, which was attributed to 120 mg NPJ5008: the muscle weakness intensity improved to moderate in less than 1.5 h without intervention and resolved within 24 h. The majority of TEAEs were mild, and most were classified as adverse drug reactions (ADR). One participant receiving Dantrium®, and two participants receiving 60 mg NPJ5008, experienced moderate muscle weakness, and of these, one also experienced a moderate headache considered unrelated to NPJ5008. Four of the five participants in Part 2 experienced moderate TEAEs of muscle weakness (n = 2) and dizziness (n = 3); none required medical intervention.
Table 3 - Adverse events experienced by more than 10% of participants, coded according to preferred terma Study Part 1 Study Part 2 Dantrium® 60 mg (N = 16) NPJ5008 60 mg (N = 16) NPJ5008 120 mg (N = 5) Adverse event (AE) n (%) Total number of events n (%) Total number of events n (%) Total number of events AEb (TEAE) 15 (93.8) 49 16 (100) 61 5 (100) 35 Serious TEAE 0 0 0 0 0 0 Severe TEAE 0 0 0 0 1 (20) 1c (one muscular weakness event) Moderate TEAE 1 (6.3) 1c (one muscular weakness) 2 (12.5) 3c (one headache; two muscular weakness) 3 (60) 5c (two muscular weakness; three dizziness) Adverse drug reactionsd (ADR) 15 (93.8) 45 16 (100) 56 5 (100) 35 Dizziness 14 (87.5) 14 14 (87.5) 14 5 (100) 8 Muscular weakness 9 (56.3) 9 8 (50.0) 8 5 (100) 8 Dysarthria 6 (37.5) 6 6 (37.5) 6 5 (100) 5 Blurred vision 6 (37.5) 7 9 (56.3) 10 4 (80) 4 Dyspnoea 2 (12.5) 2 3 (18.8) 3 2 (40) 2 Somnolence 2 (12.5) 2 1 (6.3) 1 1 (20) 1 Headache 0 0 2 (12.5) 3 1 (20) 1 Dysphagia 0 0 0 0 3 (60) 3 Balance disorder 0 0 2 (12.5) 2 0 0 Infusion site discomfort 1 (6.3) 1 1 (6.3) 1 0 0 Nausea 0 0 1 (6.3) 1 1 (20) 1 Flushing 0 0 2 (12.5) 2 0 0 Dysphonia 0 0 0 0 1 (20) 1Mild adverse events of catheter site-related reactions, flatulence, abdominal discomfort and feeling cold were considered unrelated to the study drugs. N is the total number of participants in the group, n is the number of participants reporting at least one event, % is percentage of total participants in group.
留言 (0)