
記住我
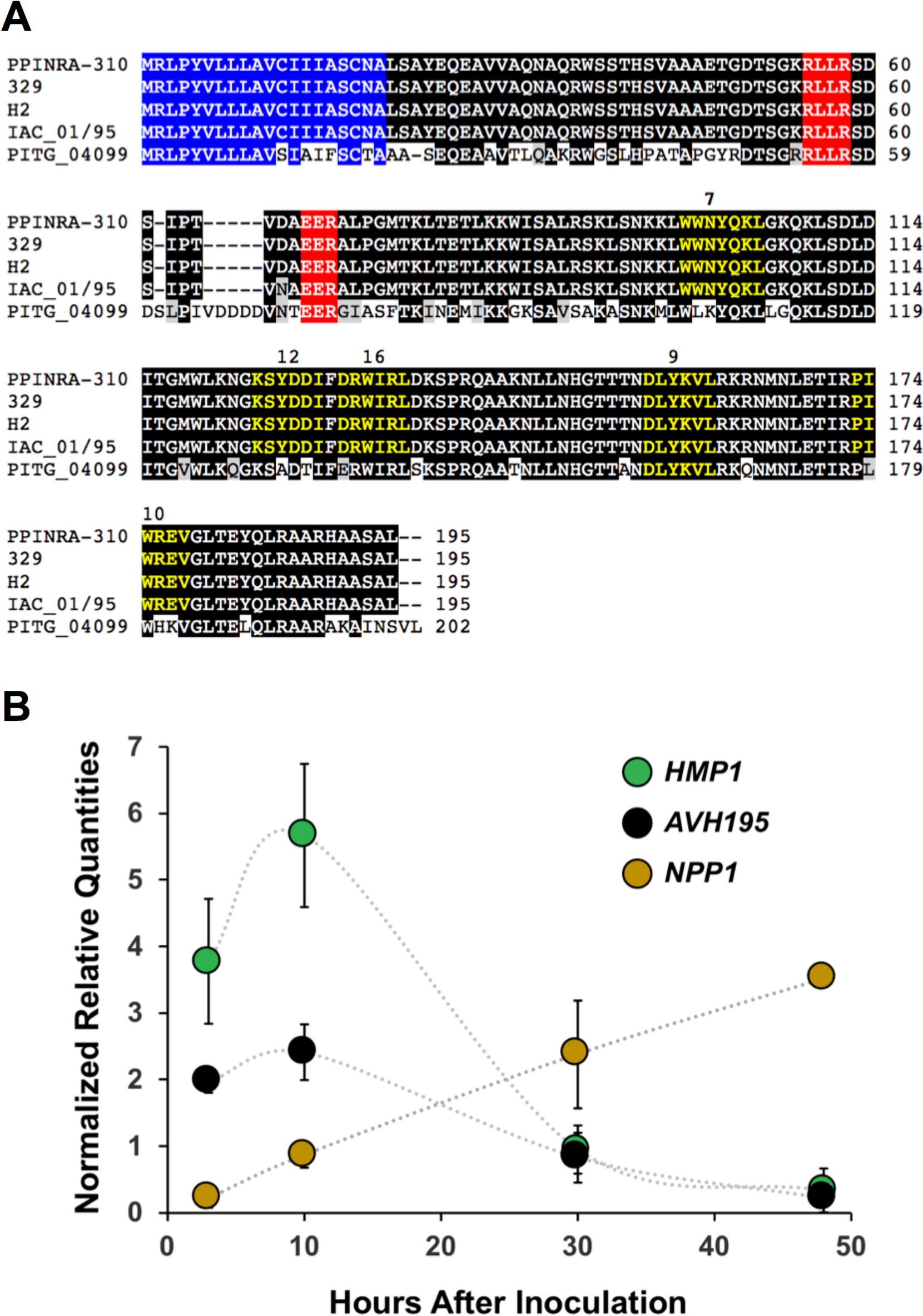
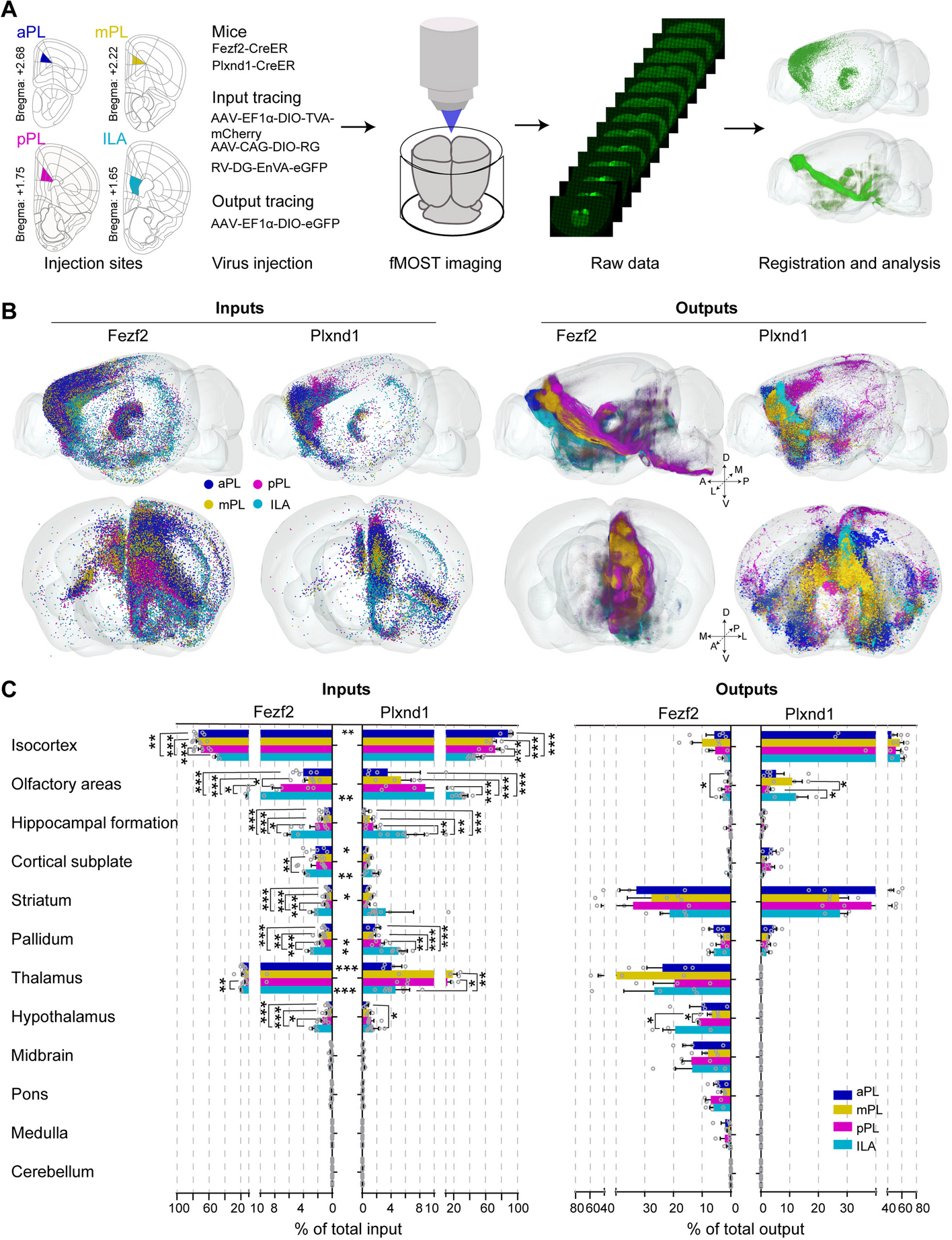

Following scRNA-seq of induced cell types at different times and co-culture conditions, we performed read alignment, tabulated gene level counts per million (CPM), calculated log2(CPM + 1), and performed principal component analysis (PCA) followed by Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction followed by K-nearest neighbor (KNN) graph construction in PC space and cell clustering with Louvain optimization of modularity [12] within the Seurat package in the R/Bioconductor environment to explore cells and homogeneity within cell types (see the “Methods” section for details). The hiPSC-N15, hiPSC-N21, and hiPSC-Ast0, which were grown in separate plates from each other, showed that, as expected, hiPSC-N and hiPSC-A clustered separately (Fig. 1A). This allowed us to distinguish hiPSC-N21A from hiPSC-AN21 based on their transcriptome profile despite them being grown in the same plate. We then calculated pair-wise correlations (r2) of pseudobulk expression profiles of all genes across the 5 conditions: hiPSC-N15, hiPSC-N21, hiPSC-N21A, hiPSC-A0, and hiPSC-AN21 (Fig. 1B). Within target cell types (i.e., within hiPCS-N or hiPCS-A), the correlations between the different conditions were strong (min r2 > 0.96), while across different discrete cell types, they were significantly weaker (max r2 < 0.58) highlighting the difference between hiPSC-N and hiPSC-A. To determine how faithfully each iPSC-derived group represents in vivo excitatory neurons and astrocytes, we explored the expression of a long list of astrocytic and neuronal marker genes compiled based on what is commonly seen in the literature and our own experience. As expected, hiPSC-A0 and hiPSC-AN21 exhibited higher expression of astrocytic markers than neurons, and this was more pronounced in the hiPSC-AN21 which may be an indication of higher maturity. The fraction of the cells in each cell type expressing the genes in Table 1 and the mean expression of each gene in each cell type are shown in Additional files 1, 2, 3, 4 and 5: Sup. Figure 1, Sup. Figure 2, Sup. Figure 3, Sup. Figure 4, and Sup. Figure 5.
Fig. 1
UMAP plot of our data highlighting the different conditions (A), pairwise correlations of gene expression in bulk and pseudobulk gene expression for the conditions (B), the same UMAP plot highlighting cluster derived with Louvain clustering (C), and compositions of the Louvain clusters in terms of cells from the five conditions (D)
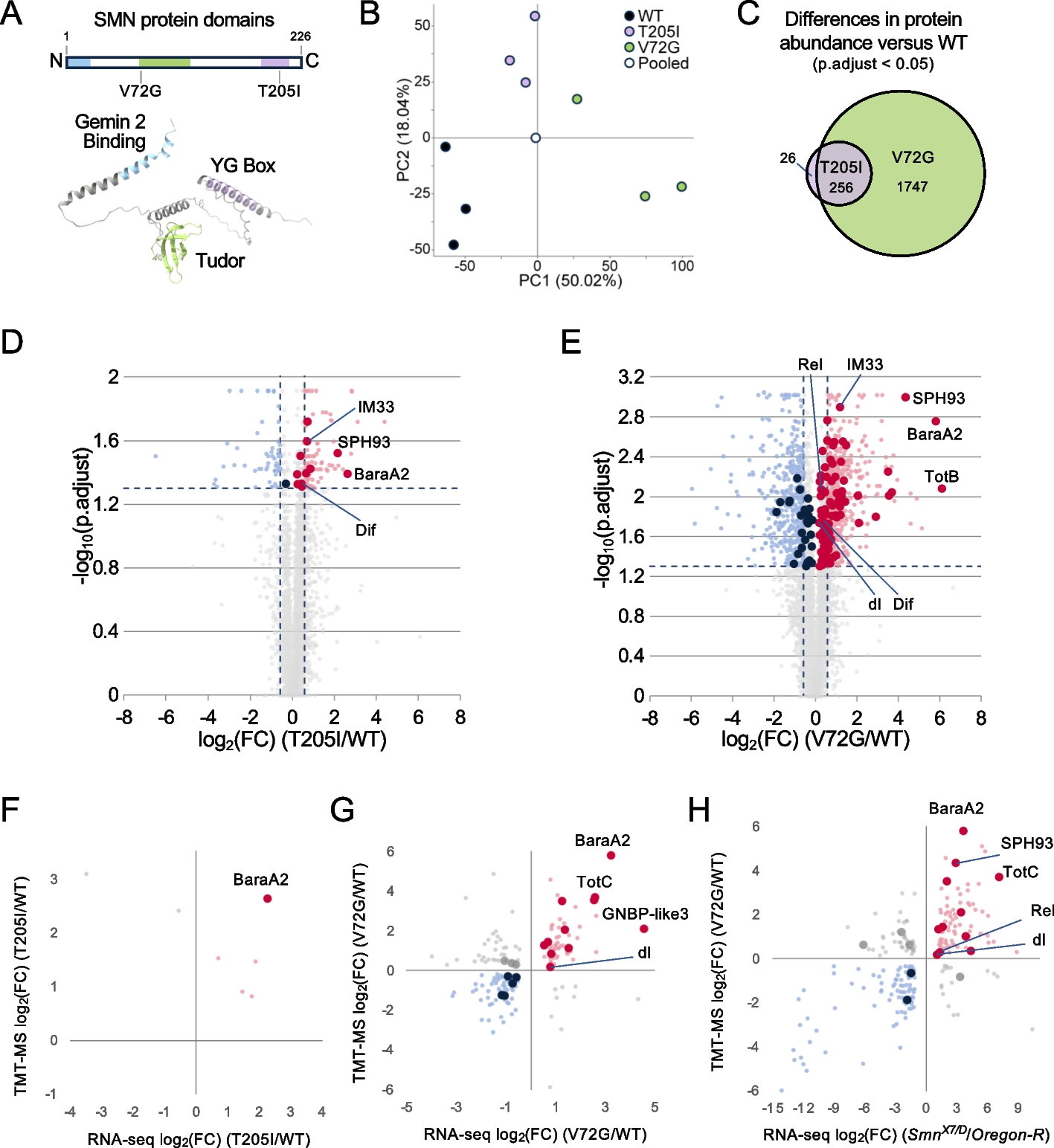
Table 1 Expression of marker genes across the different conditions. Overall expression in counts per million is shown in column 3, with darker grey shading indicating higher expression. The following columns show the expression in each condition expressed in standard deviations from the mean and proportionally highlighted from blue (negative) to red (positive). Asterisks indicate statistical significance < 0.05 between adjacent columns, with the column named A-N-SIG indicating statistical significance < 0.05 between all neuronal and al astrocytic cells typesFig. 2
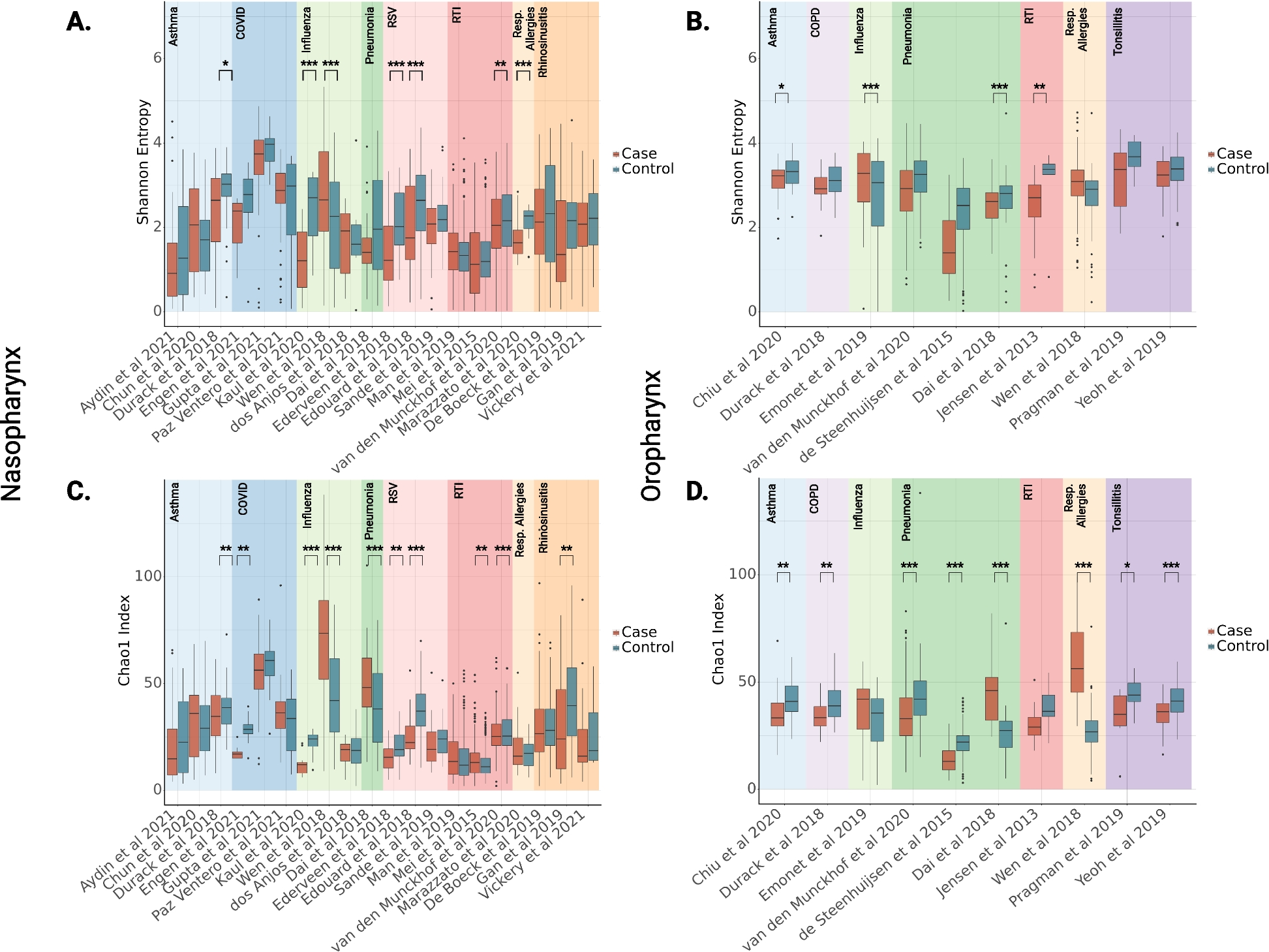
A MetaNeighbor analysis of our bulk and pseudobulk expression data with in vivo data from two in vivo studies. Ex_Cor, excitatory cortical; HEW, human embryo week; Astro, astrocytes; Oligo, oligodendrocytes; OPC, oligodendrocyte precursor cells; Endo, endothelial. B, C Seurat integration analysis of our hiPSC-N cells with neuronal cells from two in vivo datasets. B our cells colored by condition. C our cells colored by Louvain cluster. Ex_Cor, excitatory cortical; HEW, human embryo week. D, E Seurat integration analysis of our hiPSC-A cells with non-neuronal cell from two in vivo datasets. D Our cells colored by condition. E Our cells colored by Louvain cluster. SMC, smooth muscle cells; VEC, vascular endothelial cells; VLMC, vascular leptomeningeal cells. Astro, astrocytes; Oligo, oligodendrocytes
Fig. 3
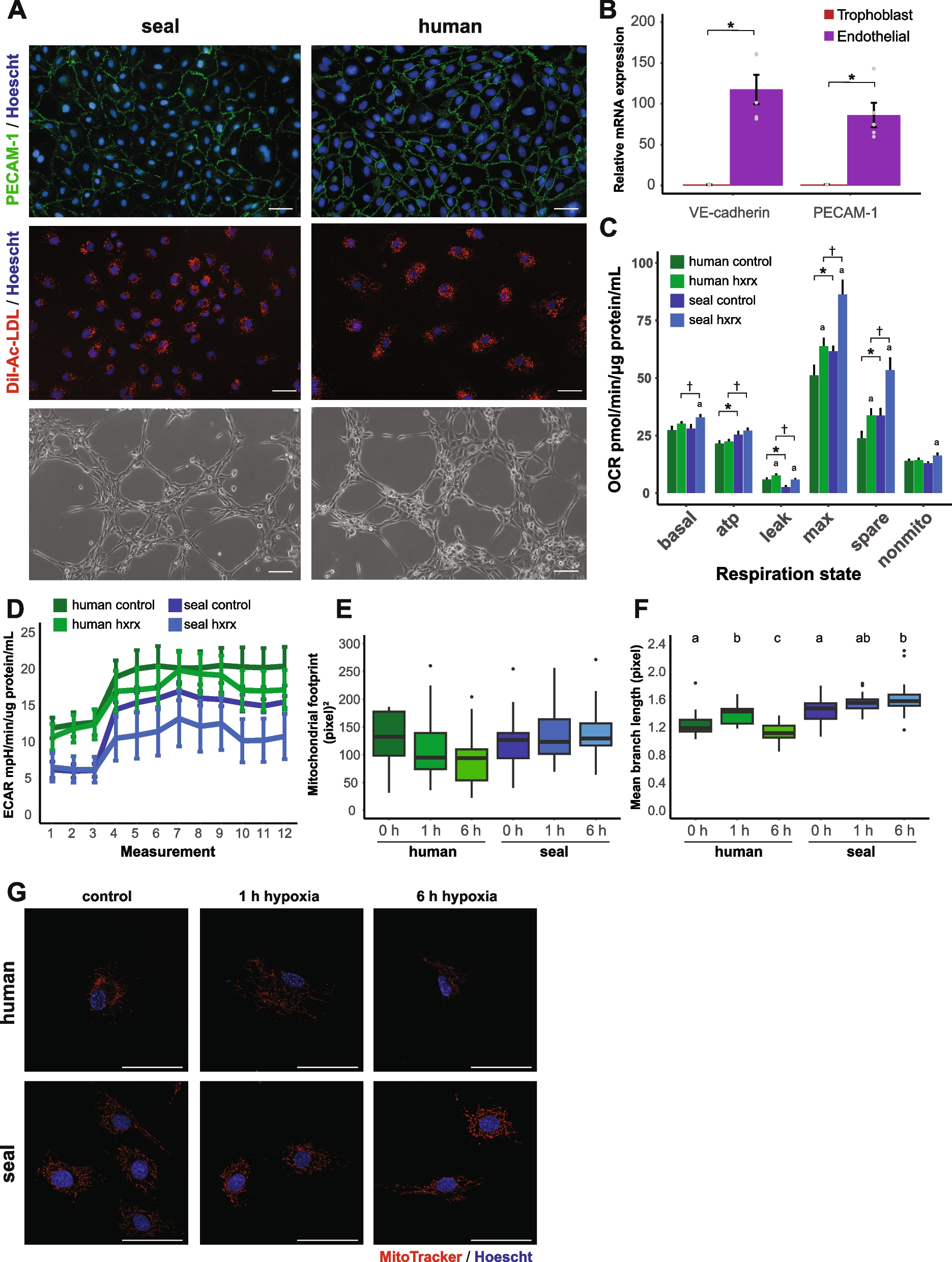
Integration analysis of our Ngn2-induced neuronal data with other single-cell data in Ngn2-induced neurons. A UMAP of Seurat-integrated data from our study and two other scRNA-seq studies of Ngn2-induced neurons (Schornig et al. and Lin et al.), colored by cell cluster. B Same UMAP colored by weeks of Ngn2 induction. C Proportion of cells from different time points in each cell cluster. D Cells from individual studies visualized in the same UMAP as in A and B, again colored by weeks of Ngn2-induction. E Expression of marker genes used to delineate diversity in the lineage composition of Ngn2-indced neurons. F MetaNeighbor analysis of in vitro cells and in vivo cell types (both divided by time points and by study: W, weeks of Ngn2 induction or gestational week; HE, human embryo, individual cell lines used are also indicated)
Fig. 4
Correlation matrix of genome-wide pseudo-bulk expression data from hiPSC-A cells, bulk RNA-seq data from additional in vitro iPSC-derived astrocytes (Tcw = Brennand) and in vivo cell types (Fan and Darmanis). iAstro1-4, in vitro iPSC-derived astrocytes; CtxAstro & MbAstro, cultured primary cortical and midbrain astrocytes. Only the 3000 most highly variable genes were used in the calculation of the correlation coefficients
Most neuronal marker genes showed higher expression in hiPSC-N than hiPSC-A with one exception, SATB2. SATB2 is a postmitotic determinant for upper-layer neuron specification not present in all neurons [13]. While this can explain its absence in hiPSC-N, it is not clear why we observe it expressed in hiPSC-A. Multiple calcium/calmodulin-dependent protein kinases (CaMK) were highly expressed in hiPSC-N. The only exception was CAMK2D whose expression was higher in hiPSC-A. This is in agreement with Vallano et al. [14] who have shown that CAMK2D is a CaM kinase type II with specific astrocyte expression. All other synaptic markers showed high expression in hiPSC-N, often with a significant trend for higher expression in the direction hiPSC-N15➔hiPSC-N21➔hiPSC-N21A, like the glutamatergic markers GRIA1 and GRIN3A and the synaptic gene NRXN3 and CAM2N2. This positive correlation of glutamatergic and synaptic gene expression with time post-Ngn2 induction and co-culture with hiPSC-A may suggest increasing maturation of the neurons across time and co-culture.
We next wanted to explore whether our differentiated cells, all of which come from a single cell line, are different or similar in expression profiles with cells derived from different cell lines. We compared our hiPSC-N to others we differentiated following the same protocol in our previous work [15,16,17,18] and with an in vivo dataset [19, 20] (Additional file 6: Sup. Figure 6). Similarly, we explored whether our astrocytes are similar with astrocytes differentiated by others with the same protocol or in vivo fetal and mature astrocytes [8, 21,22,23,24,25,26,27] (Additional file 7: Sup. Figure 7). With the neuronal cells we found strong correlations (r > 0.8) with all the bulk in vitro datasets and r > 0.7 with the in vivo bulk dataset (Additional file 6: Sup. Figure 6). With the cells differentiated to astrocytes, we also found strong correlations (r > 0.74) with all the bulk in vitro datasets and r > 0.49 with the in vivo bulk datasets (Additional file 7: Sup. Figure 7). This suggests that the genomic differences between cells and other unintentional differences in experimental procedures do not have a major effect on the transcriptomic signature of the cells.
We next harnessed the power of single-cell sequencing to explore the cellular homogeneity of these hiPSC-derived differentiated cells. Instead of specifying cell groups based on the culture condition (or visualization in the case of hiPSC-N21A vs. hiPSC- A N21) as above, we applied Louvain community detection, a method to extract communities with shared features from large networks [12], which identified 8 clusters within our cells (Fig. 1C). Five clusters included exclusively derived neurons—N-I to N-V—and 3 included derived astrocytes—A-I, II, and III. None of them is composed of a single condition (Fig. 1D). This suggests they are influenced by but do not solely depend on the different conditions and likely reflect a property of the base differentiation methods. Cluster A-III showed a biased composition by condition, containing > 90% hiPSC-AN21. Comparing gene expression levels of genes in each neuronal cluster to the remaining 4 neuronal clusters identified numerous genes, differentially expressed. These, along with the astrocytic cluster comparisons, can be found in Additional file 8: SuppTable1. According to the statistical overrepresentation test of the PANTHER bioinformatics tool [28], they were functionally enriched for many neural development and synaptic genes compared with the whole set of genes expressed in the 5 clusters (see Additional file 9: SuppTable2 for results on each cluster). Most enrichments were observed among genes expressed lower in examined cluster, while some were in both upregulated and downregulated genes. Similarly, comparing gene expression levels of genes in each astrocytic cluster compared with the remaining 2 identified numerous genes with enrichments compared with the whole set of genes expressed in the 3 clusters, in this case only in genes expressed lower in the examined cluster. These also included, among others, neuron-related functions (Additional file: 10: SuppTable3).
In summary, our observations support that iPSC-N and iPSC-A express marker genes that broadly suggest similarity to in vivo excitatory neurons and astrocytes as previous studies of these cells in bulk have shown [7, 8]. However, we do find heterogeneity which is influenced by the specific conditions of differentiation that we tested. This suggests that the heterogeneity is in part inherent to the differentiation protocols. Heterogeneity aside, the gene marker data suggests a role of the presence of neurons in the maturation of astrocytes and vice versa. Similarly, differentiation time also seems to play an important role for the maturation of the neurons.
GWAS genes expressed in iPSC-derived neurons and astrocytesTo be appropriate disease models, the cells created by these differentiation methods must express many of the genes associated with diseases involving neurons and astrocytes. We compared the genes showing differential expression (DE) between hiPSC-N and hiPSC-A (hiPSC-N and hiPSC-A specific genes) to those associated with neuropsychiatric illness by large genome-wide association studies (GWAS) and sequencing studies. We focused on schizophrenia (SZ) and Alzheimer’s disease (AD) due to the availability of large GWAS [29, 30] and autism spectrum disorders (ASD) where large sequencing studies from the Simons Foundation Autism Research Initiative have identified many genes [31]. In the case of ASD, the data is from sequencing studies identifying clustering of damaging variants, providing a direct link to specific genes. In the case of SZ, the genes were reported as high confidence based on co-localization with brain eQTLs and posterior probability analyses [29]. Similarly, the AD GWAS assigned genes to associated variants based on colocalization analysis, fine-mapping results, and previous literature [30]. Using DESeq2 for differential expression analysis, we used a stringent threshold of an adjusted p < 0.001 (see the “Methods” section) to focus on the genes with the highest confidence of DE between cell types. We also used the highest confidence genes reported for each disorder as follows: for ASD, this was 207 genes with a score of 1 (highest confidence) in the SFARI database; for SZ, this was 130 genes reported as genome-wide significant with high confidence [29]; for AD, this was 38 genes at loci showing genome-wide significant association [30].
Of the 15,157 genes in our dataset, 5154 were higher in hiPSC-N and 4107 in hiPSC-A at FDR < 0.001. From the 28 AD-associated genes present in our dataset, 7 were among the 5154 genes higher in hiPSC-N and 12 among the 4107 higher in hiPSC-A. For the genes higher in hiPSC-A, this is 1.6-fold more than expected (hypergeometric p = 0.022), while for those higher in hiPSC-N, it was 1.4-fold less than expected and also not significant. Out of 106 SZ-associated genes in our dataset, 56 were among those higher in hiPSC-N and 26 among those expressed higher in hiPSC-A. This is 1.55-fold more than expected for genes higher in hiPSC-N (hypergeometric p = 2.1 × 10−5) and as expected by chance for genes higher in hiPSC-A. Finally, out of 199 ASD genes in our dataset, 109 were among the genes higher in hiPSC-N and 39 among those higher in hiPSC-A, which is a 1.6-fold excess for hiPSC-N (hypergeometric p = 5 × 10−10) and a significant depletion (1.4-fold) for hiPSC-A (hypergeometric p = 5 × 10−6). These results are consistent with what is currently believed for these disorders; SZ and ASD have been genetically linked to neuronal functions [29, 32], while astrocytes have been implicated in AD [33]. The result also supports that these hiPSC-derived cells, while not equivalent to in vivo neurons and astrocytes, may be useful for modeling disease. The complete set of genes with their expression in neurons and astrocytes and the comparison of the two is in Additional file 11: SuppTable4.
Differences between hiPSC-N15 and hiPSC-N21To determine the importance of differentiation under specific conditions and its possible relevance to disease genes, we also performed DE analysis between conditions. The hiPSC-N15 are a deviation from the original Ngn2 induction protocol which reported mature neurons at 21 days post-induction [7]. While we have not formally measured the differences, we have empirically observed little morphological change after day 15, so we decided to use the transcriptome to explore how hiPSC-N15 differ from hiPSC-N21. Shorter differentiation time not only has practical advantages, but there is a possibility that it may resemble an earlier developmental time (as supported by Table 1) perhaps more important to some diseases. The complete DE analysis results for all genes are in Additional file 12: SuppTable5.
At adjusted p < 0.1, 571 of 14,095 genes were expressed higher in hiPSC-N21 and 889 higher in hiPSC-N15. Using the multiple testing corrected statistical overrepresentation test of the PANTHER bioinformatics tool [28] which allows comparisons to user-provided reference lists (in this case the list of all 14,095 genes) and biological processes annotations from the gene ontology database [34], we found among the genes expressed higher in hiPSC-N15 significant enrichments (adjusted p < 0.05) for terms including “neurogenesis,” “neuron projection morphogenesis,” “cell morphogenesis involved in neuron differentiation,” and “regulation of neuron projection development” (see Additional file 13: Sup. Figure 8). Among the genes expressed higher in hiPSC-N21, we found significant enrichments for the GO terms “negative regulation of neuron death,” “regulation of neurotransmitter levels,” “chemical synaptic transmission,” “neuron differentiation,” and “nervous system process” (see Additional file 14: Sup. Figure 9). This suggests that genes in early neuronal development are expressed higher in hiPSC-N15, while genes involved in neuronal function are higher in hiPSC-N21 and that hiPSC-N15 are less mature compared with hiPSC-N21. This may suggest that, despite the artificial course of differentiation, hiPSC-N15 may more closely resemble neurons earlier in their course to maturity.
To gain insight into the importance of these genes in neurodevelopmental disorders, we intersected them with the 130 genes reported by the Psychiatric Genomics Consortium (PGC3 data) [29] as associated with SZ with highest confidence. Of these, 105 were present in the expressed gene list, and of those, 18 (17%) were significantly higher (7) or lower (11) in hiPSC-N21 (Additional file 15: SuppTable6). While this is not significant for each direction separately (each (hypergeometric p ~ 0.06), it is significantly (1.7-fold) more DE genes than expected by chance (hypergeometric p = 0.01). We further compared them to the list of 207 genes reported as high confidence for ASD by SFARI. Of these, 199 were present in our expressed gene list, and 12 were expressed higher in hiPSC-N21 (hypergeometric p = 0.06) and 22 lower (hypergeometric p = 0.003). Overall, there were 34 DE genes in either direction 1.65-fold more than expected by chance (hypergeometric p = 0.001) (Additional file 15: SuppTable6), observing that DE genes in both directions appear to be related to risk for ASD, and SZ is complicating the choice of differentiation time for modeling disease.
Differences between hiPSC-N21 and hiPSC-N21AWe then explored how co-culture with hiPSC-A affects the transcriptome of hiPSC-N. The complete transcriptome comparison results for all genes are in Additional file 16: SuppTable7. Since hiPSC-N21A neurons were grown in the same plate as hiPSC-AN21 astrocytes, to avoid artifacts due to possible contribution of cell-free RNA (also termed “ambient RNA”) from lysed cells in the co-culture media that could be sequenced and confound results [35], we excluded from this comparison genes that were expressed markedly higher in hiPSC-A than hiPSC-N at adjusted p < 0.001(“high hiPSC-A genes”). Note that Additional file 16: SuppTable7 contains all results for all genes detected, with those excluded marked accordingly.
Overall, the expression of 359 out of 11,222 genes included in the analysis was higher in hiPSC-N21A than hiPSC-N21 at adjusted p < 0.1. PANTHER bioinformatics showed a 3.3-fold enrichment for genes involved in “regulation of metal ion transport” and 1.5-fold enrichments for “cell communication,” “signal transduction,” and “signaling” (Additional file 17: Sup. Figure 10). Five hundred ten genes were expressed significantly higher in the absence of astrocytes (hiPSC-N21) at adjusted p < 0.1 out of 14,857 included in the analysis. There was a 7.1-fold enrichment for “central nervous system neuron axonogenesis,” a 3.2-fold enrichment for “axon guidance,” 2.8-fold for “axon development, 2.1-fold for neuron development, and 1.6-fold for “cell differentiation” (complete results in Additional file 18: Sup. Figure 11). The enrichments for axon development and guidance and neuron development, including genes like SEMA4D and DCC [36], the CRMP5-encoding DPYSL5 [37], and the SLIT-ROBO Rho GTPase-activating protein SRGAP1 [38], suggest an earlier stage of development for hiPSC-N21 and support the importance of the inclusion of astrocytes in maturation.
We compared these genes to the list of 130 high confidence SZ-associated genes reported by the PGC [29]. In contrast to the overlap with genes differing between hiPSC-N15 and hiPSC-N21, here, only 5 of these genes were among those higher in hiPSC-N21A (MSI2, CUL9, CSMD1, OPCML, and DCC) and 4 among those higher in hiPSC-N21 (MAPK3, NXPH1, IL1RAPL1, GALNT17). Similarly, when compared with the ASD genes, only 8 were among those higher in hiPSC-N21A and 11 among those higher in hiPSC-N21, not significantly more than expected. This could suggest that disease genes might not be among those impacted by the co-culture of neurons and astrocytes or could be due to reduced power.
Differences between hiPSC-A0 and hiPSC-AN21We next explored how co-culture with hiPSC-N affects the transcriptome of hiPSC-AN. The complete transcriptome comparison results for all genes are in Additional file 19: SuppTable8. Similarly to the reverse comparison, to avoid confounding from cell-free RNA from hiPSC-AN21 lysed cells to droplets containing hiPSC-N21A, we excluded genes that were significantly higher in hiPSC-N compared with hiPSC-A (at adjusted p < 0.001). Note that our Additional file 19: SuppTable8 contains all results for all genes detected, with those excluded marked accordingly.
In all, out of 10,237 genes included in the analysis after the removal of the “high hiPSC-N” genes, 583 were significantly higher in hiPSC-AN21 than hiPSC-A0 at adjusted p < 0.1, and PANTHER bioinformatics showed multiple significant functional enrichments (Additional file 20: Sup. Figure 12). Notably, we observed 2.3- to ninefold enrichments for functions including “regulation of superoxide metabolic process,” “cellular oxidant detoxification,” and “response to oxidative stress,” all important functions of astrocytes in their supportive roles nervous system [39]. Due to the importance of astrocytes in AD [40], we also looked for AD GWAS genes for enrichments. Twenty-one AD-associated genes were present in the reference list of genes in our comparison, and 3 of them were among the 583 significantly higher in hiPSC-AN21 (APOE, CLU, and CASS4), compared with 1.2 expected by chance (hypergeometric p = 0.02). While this is a small number of genes, it is important to know that studying them using in vitro differentiated astrocytes might benefit from the inclusion of neurons in the cultures. This is particularly important for APOE which is a very widely studied AD gene.
When it comes to genes that were higher in hiPSC-A0 than hiPSC-AN21 out of 14,912, there were 1071 at adjusted p < 0.1, and PANTHER bioinformatics analysis also showed multiple significant functional enrichments, shown in Additional file 21: SuppTable9, and those with more than threefold enrichments are also illustrated in Additional file 22: Sup. Figure 13. Most striking were more than eightfold enrichments for immunity related genes. Of those directly relevant to neural cells, there were multiple categories involving neuron development and neural tube closure as well as axon development and guidance.
Among 27 AD-associated genes in the tested set of genes, there were 4 AD-associated genes higher in hiPSC-A0 vs. hiPSC-AN21 (GRN, PICALM, APH1B, and CD2AP), a 2.1-fold excess from expected (hypergeometric p = 0.04). We observe again excess occurrence of disease genes on both sides of the distribution, suggesting that the better choice of cells for disease modeling might be gene specific.
The top 50 genes in each cell type contrast, detailed in the “Differences between hiPSC-N15 and hiPSC-N21,” “Differences between hiPSC-N21 and hiPSC-N21A,” and “Differences between hiPSC-A0 and hiPSC-AN21” sections, are shown in heatmaps in Additional file 23: Sup. Figure 14. In order to further explore the similarity of these in vitro cells to multiple in vivo lineages, we compare the expression data for the top differentially expressed genes in our in vitro derived cells to data from additional studies of related in vivo cell types [41,42,43,44,45,46] and to directed differentiation in vitro [47, 48].
Comparisons with in vivo datasetsHaving used individual a priori known marker genes and differential expression to show that hiPSC-N and hiPSC-A express many of the expected markers for their intended cell type but also show heterogeneity within cell type, we then compared these cell types and their subclusters to in vivo cells in two previously reported datasets by Darmanis et al. [41, 44] and Fan et al. [19, 20].
Darmanis et al. [41] performed single-cell transcriptome analysis on adult and human fetal brain. Fan et al. [41] performed single-cell transcriptome profiling of cells from the four cortical lobes and pons during human fetal development from the 7th to the 28th gestational week (GW). We first performed a MetaNeighbor analysis [49] across all genes in the three datasets to explore similarities across cell types in the different experiments. This analysis is based on a statistical framework that quantifies the degree to which cell types replicate across datasets [49]. The complete heatmap is shown in Additional file 24: Sup. Figure 15, while Fig. 2A includes the portion comparing our in vitro derived cells to the in vivo cell types. As expected, the Ngn2-induced neurons are closest to the in vivo neuronal cell types, includi
留言 (0)