
記住我



Regorafenib is an oral multikinase inhibitor that targets several receptor tyrosine kinases such as vascular endothelial growth factor receptor, platelet-derived growth factor receptor beta, fibroblast growth factor receptor 1, c-Kit, RET, and intracellular signaling kinases such as RAF [1]. Regorafenib has been shown to inhibit tumor angiogenesis, tumor cell proliferation, and mitogen-activated protein kinase signaling in xenograft models of melanoma, colorectal, breast, renal cell, pancreatic, and lung cancers [2]. Regorafenib is currently approved by the Food and Drug Administration (FDA) for use in metastatic colorectal cancer, hepatocellular carcinoma (HCC), and advanced gastrointestinal stromal tumors (GIST) [3–5].
Sildenafil citrate is an oral phosphodiesterase-5 (PDE5) inhibitor approved by the FDA for the treatment of erectile dysfunction [6] and pulmonary hypertension [7]. PDE5 hydrolyzes the 3’,5’ PDE bond in cyclic GMP (cGMP) [8]. PDE5 is a major PDE isoform overexpressed in human primary breast tumors [9], pancreatic tumors [10], lung carcinomas [11], and colon cancers [12]. By inhibiting PDE5, sildenafil increases intracellular cyclic GMP (cGMP) leading to nitric oxide-mediated vasodilation in diverse tissues [7,8]. It has been shown that cGMP exerts antiproliferative and proapoptotic effects [13,14]. Modulation of cGMP levels by sildenafil in tumor cells may promote the ability of tumor cells to undergo apoptosis in response to chemotherapeutic agents. This effect has been seen using a wide array of cytotoxic and biologic agents (including trastuzumab, gemcitabine, doxorubicin, cisplatin, oxaliplatin, mitomycin C, plant polyphenols, and paclitaxel) in acute myeloid leukemia, and bladder, lung, breast, and pancreatic cancers [15–18].
We have previously shown that clinically relevant concentrations of regorafenib interact with sildenafil in a synergistic fashion to kill multiple cancer cell types, both in short-term assays and in colony formation assays [19].
Based on data from several clinical trials, including the Regorafenib Monotherapy for Previously Treated Metastatic Colorectal Cancer (CORRECT) trial [3,4,20–22], the suggested and FDA-approved dose for regorafenib is 160 mg orally once daily for the first 3 weeks of a 4-week cycle. The recommended dose for sildenafil varies from 25-100 mg once daily based on the indication for erectile dysfunction. For patients using sildenafil for pulmonary hypertension, a dose of 80 mg 3 times per day was well tolerated [23].
While the regorafenib and sildenafil combination had not previously been evaluated in a clinical setting, one study evaluated the use of sorafenib, a multikinase inhibitor with toxicity similar to that of regorafenib, in patients receiving sildenafil for pulmonary arterial hypertension [24]. This combination was safely administered without any grade 3 or 4 toxicities.
Materials and methods Drug supplyRegorafenib was provided by Bayer HealthCare Pharmaceuticals, Inc. Sildenafil commercial stock was obtained by VCU MCC and provided at no charge to study participants. Both drugs were provided through the VCU Health System Investigational Drug Service.
Patient eligibilityPatients 18 years of age or older with an ECOG performance status of 0 or 1 and an advanced solid tumor that progressed during or after treatment with approved therapies, or for which no standard effective therapy was available, were eligible for this study. Adequate bone marrow (as measured by white blood cell and platelet counts and hemoglobin levels), renal, and hepatic function was required. Patients with symptomatic or untreated meningeal or brain metastases were excluded. Use of investigational agents within 4 weeks before initiating study treatment, or any previous therapy with regorafenib, was prohibited. Ineligible patients also included those with an intolerance to sorafenib, inability to swallow medication, known or suspected malabsorption condition or obstruction, contraindications to sildenafil (including known retinitis pigmentosa, history of priapism related to PDE5 inhibitors, or presence of nonmalignant hematologic disorders), contraindication to antiangiogenic agents, history of organ allograft, history of bleeding or blood vessel problems, low resting systolic blood pressure, hypertension despite optimal medical management, cardiac disease, seizure disorder, serious uncontrolled infection, known HIV infection, chronic or active hepatitis B or C infection, pleural effusion or ascites causing respiratory compromise, untreated or metastatic pheochromocytoma, or planned ongoing treatment with other drugs thought to potentially have adverse interactions with either regorafenib or sildenafil.
Treatment planPatients took both regorafenib and sildenafil once daily at the same time on days 1 through 21 of a 28-day cycle. No regorafenib or sildenafil was taken on days 22 through 28 of the cycle. Treatment continued until disease progression or development of unacceptable toxicity. Treatment was discontinued due to unresolved toxicity lasting for more than 28 consecutive days or if the patient or investigator determined discontinuation was in the patient’s best interest. Patients were instructed to take any missed dose as soon as they remembered on the same day and not to take 2 doses of either drug on the same day to make up for doses missed on the previous day.
Study designThis study was a phase 1, single-center, open-label, single-arm, dose-escalation trial using a 3 + 3 design. Within each dose level (Table 1), 3–6 patients were enrolled. Additional patients were enrolled at the maximum tolerated dose (MTD) until a total of 12 patients were treated at the MTD. The suggested dose and FDA-approved dose for regorafenib is 160 mg orally once daily for the first 3 weeks of a 4-week cycle. However, given that 67% of patients required regorafenib dose modifications in the CORRECT trial [3], the starting dose of regorafenib dose for the first cohort was 120 mg given with sildenafil 50 mg once daily for the first 3 weeks of each 4-week cycle. If no dose-limiting toxicity (DLT) was observed, the regorafenib dose was increased to 160 mg for the next cohort. This study was reviewed and approved by the Institutional Review Board of Virginia Commonwealth University and was performed according to the principles defined by the Declaration of Helsinki.
Table 1 - Regorafenib and sildenafil dose levels and dose-limiting toxicities Dose level Regorafenib Sildenafil Patients treated DLT evaluable DLT events DLT event details Taken orally once each day on days 1 through 21 1 120 mg 50 mg 4 3 0 2 160 mg 50 mg 9 6 1 Grade 4 Lipase Increased 3Aa 160 mg 100 mg 16 12 1 Grade 3 Maculo-papular Rash & Grade 3 Pain 3B 120 mg 100 mg 0 0 0aMaximum tolerated dose (MTD).
The primary objective was to determine the recommended phase 2 dose of the combination of regorafenib and sildenafil in patients with advanced solid tumors. Secondary objectives included identification of antitumor effects of the regorafenib and sildenafil combination, evaluation of toxicity of the regorafenib and sildenafil combination, determination of pre-treatment expression of PDE5 in tumor samples, and evaluation of the impact of sildenafil on the pharmacokinetics of regorafenib.
EvaluationAll adverse events were collected and evaluated using NCI CTCAE version 4. Tumor response was evaluated according to RECIST v1.1 [25].
Dose-limiting toxicity evaluabilityDose-limiting toxicities (DLT) were evaluated in the first 28-day cycle. Patients must have received 75% of the assigned total doses of each drug to be considered evaluable for DLT. Any grade 3 or higher toxicity was considered to be a DLT, with the exception of the following grade 3 toxicities: nausea, vomiting, diarrhea, or fatigue were excluded only when responsive to medical management; anemia; febrile neutropenia; decrease in platelet, white blood cell, neutrophil, or lymphocyte counts; and the first incidence of palmar-plantar erythrodysesthesia. Additionally, grade 4 decrease in lymphocyte count was not considered a DLT. Hematological changes are often seen in patients with advanced cancer as a result of their disease state, and don’t necessarily impact their ability to receive therapy safely. Thus, grade <3 hematological toxicities (and grade <4 decreases in lymphocyte count) were excluded as DLTs as we chose to focus on toxicities that were dangerous enough to preclude administration of therapy to other patients. Any hematological toxicity was considered an SAE if it resulted in a hospital visit.
Safety evaluabilityPatients who received any study treatment were included in evaluations of safety, toxicity, and tolerability.
Efficacy evaluabilityPatients who received any doses of regorafenib in cycle 2, regardless of the number of doses taken during cycle 1, and had at least 1 imaging assessment of response after receiving study treatment were included in evaluations of treatment efficacy.
Pharmacokinetic and pharmacodynamic analysis Multiplex analysisWhole blood (1 mL) was collected in EDTA blood collection tubes at the following timepoints: (1) day 1: 3 h after initial dose of regorafenib and sildenafil, (2) day 15: before dosing; (3) day 15: 3 h after dosing; (4) day 21: before dosing; (5) day 21: 3 h after dosing. Blood samples were promptly mixed by gently inverting 8-10 times and centrifuged (500g, 10 min, 4 °C) within 30 min after collection. The plasma was separated and aliquots were stored at −80 °C.
The Human Immune Monitoring 65-plex ProcartaPlex Panel for MAGPIX (ThermoFisher) was used for the simultaneous detection and quantification of 65 protein targets in 25 µL of plasma according to the manufacturer’s instructions.
PharmacokineticsWhole blood (6 mL) was collected in EDTA blood collection tubes at the same timepoints described for the Multiplex Analysis. Samples were promptly mixed by gently inverting 8–10 times, and centrifuged (1500g, 10 min, 4 °C) within 30 min of collection. The plasma was separated and aliquots were stored at −80 °C.
Collected plasma samples were sent to NorthEast BioAnalatical Laboratories LLC (Hamden, CT) for the quantitation of regorafenib using validated method MET099 v1 titled ‘Analytical Method and Validation Report for the Determination of BAY 73-4506, BAY 75-7495 (M-2), BAY 75-1098 (M-4) and BAY 81-8752 (M-5) in Human Plasma by LC/MS/MS’ conducted in compliance with principles of Good Clinical Practice (US FDA & ICH GCP Guidance) Standards, and the Declaration of Helsinki.
ImmunohistochemistryArchived tumor tissue samples were used to determine the level of PDE5. Anti-PDE5A/PDE5 antibody ab14672 (Abcam) in 1/50 dilution was used with automated system DAKO Autostainer Plus. The results were scored by a pathologist using a scale from 2 to -1, where: 2 represents significantly stronger expression in the neoplasm compared to adjacent non-neoplastic cells; 1 represents stronger expression in neoplasm than adjacent non-neoplastic cells; 0 represents equal expression in neoplasm as compared to adjacent non-neoplastic cells; and -1 represents weaker expression in neoplasm as compared to adjacent non-neoplastic cells.
Statistical analysisDemographics, adverse events, DLTs, dose levels, and clinical responses were summarized by basic descriptive statistics, such as frequency, proportion, mean, median, and range. Due to large standard deviations and non-normality observed among the multiplex analysis responses, a natural log transformation was used to achieve close to normality. Then, a repeated measure analysis of variance (RMANOVA) was fit factoring in the correlation due to the longitudinal design using a linear mixed-effects model. The model fit included a between-subjects factor (Benefit, yes vs. no), a within-subject factor (Time, 6 levels), and an interaction term between Benefit and Time. A subject-level random effect was included to accommodate the within-subject correlation. The multiple comparison tests following the RMANOVA procedure were Bonferroni corrected. Statistical significance was determined using α = 0.05. All analyses were performed using SAS v9.4.
Results Patient characteristicsA total of 32 patients were enrolled and 29 were treated, due to eligibility screening fail (Table 2). Patients treated on trial had received 1–6 lines of prior therapy. The most common cancer types were colorectal cancer (n = 4), ovarian cancer (n = 4), pancreatic cancer (n = 3) and uterine cancer (n = 3). Patients remained on the study regimen for 12-712 days with a median treatment duration of 56 days.
Table 2 - Patient characteristics Number of patients (%)Dose levels and DLTs are reported in Table 1. DLTs were determined in Cycle 1. Of the 29 patients treated, 21 were evaluable for DLT with the remaining 8 receiving insufficient study drug within the first cycle to meet the evaluability threshold. Regorafenib is an FDA-approved agent and general dose-withholding guidelines were followed. Due to dose withholding, 8 patients were unable to complete the DLT evaluation period. There was 1 DLT at Dose Level 2 (grade 4 increased lipase), 2 DLTs at Dose Level 3A (grade 3 maculo-papular rash and grade 3 pain), and no DLTs at dose level 1. Dose Level 3A (regorafenib 160 mg, sildenafil 100 mg) was identified as the MTD.
ToxicitiesThe most common AEs attributed to regorafenib were grade 3 palmar-plantar erythrodysesthesia (n = 9), hypertension (n = 8), hypophosphatemia (n = 5), and lipase elevation (n = 3) (Table 3). One patient experienced grade 3 pancreatitis. Grade 4 hypertension was observed in 1 patient who had ovarian cancer at Dose Level 3A and grade 4 increased lipase was observed in 2 patients (1 patient with adenocarcinoma of the endocervix at Dose Level 2 and 1 patient with HCC at Dose Level 3A). No other grade 4 toxicities were attributed to regorafenib. No treatment-related deaths occurred during this study.
Table 3 - Treatment-related adverse events occurring in 20% or more of patients Adverse event category and term Number of patients n (%) All Grades Grade 1 Grade 2 Grade 3 Grade 4 Blood and lymphatic system disorders Anemia 7 (24%) 3 (10%) 3 (10%) 1 (3%) 0 Skin and subcutaneous tissue disorders Palmar-plantar erythrodysesthesia syndrome 20 (69%) 6 (21%) 5 (17%) 9 (31%) 0 Metabolism and nutrition disorders Hypophosphatemia 18 (62%) 4 (14%) 9 (31%) 5 (17%) 0 Hypocalcemia 14 (48%) 13 (45%) 1 (3%) 0 0 Hypokalemia 10 (34%) 10 (34%) 0 0 0 Anorexia 9 (31%) 5 (17%) 4 (14%) 0 0 Hypomagnesemia 6 (21%) 6 (21%) 0 0 0 Investigations Lymphocyte count decreased 14 (48%) 7 (24%) 2 (7%) 5 (17%) 0 Platelet count decreased 10 (34%) 8 (28%) 2 (7%) 0 0 Blood bilirubin increased 9 (31%) 5 (17%) 3 (10%) 1 (3%) 0 Lipase increased 9 (31%) 3 (10%) 1 (3%) 3 (10%) 2 (7%) Weight loss 8 (28%) 8 (28%) 0 0 0 Aspartate aminotransferase increased 7 (24%) 7 (24%) 0 0 0 Vascular disorders Hypertension 14 (48%) 1 (3%) 4 (14%) 8 (28%) 1 (3%) Nervous system disorders Dysgeusia 10 (34%) 7 (24%) 3 (10%) 0 0 Headache 7 (24%) 4 (14%) 3 (10%) 0 0 Renal and urinary disorders Proteinuria 10 (34%) 6 (21%) 4 (14%) 0 0 General disorders and administration site conditions Fatigue 9 (31%) 1 (3%) 6 (21%) 2 (7%) 0 Gastrointestinal disorders Diarrhea 8 (28%) 3 (10%) 5 (17%) 0 0 Nausea 6 (21%) 6 (21%) 0 0 0 Respiratory, thoracic and mediastinal disorders Hoarseness 7 (24%) 7 (24%) 0 0 0Common adverse events of sildenafil (≥2%) include headache, flushing, dyspepsia, abnormal vision, nasal congestion, back pain, myalgia, nausea, dizziness, and rash. None of our patients reported these adverse events at grade 3 or higher with probable or possible attribution to sildenafil. However, 2 patients reported grade 2 headache, 4 patients reported grade 1 headache, 2 patients reported grade 1 flushing, 2 patients reported grade 1 eye disorders (vision change, light sensitivity), 1 patient reported grade 1 back pain, and 1 patient reported grade 1 nausea with possible or probable attribution to sildenafil.
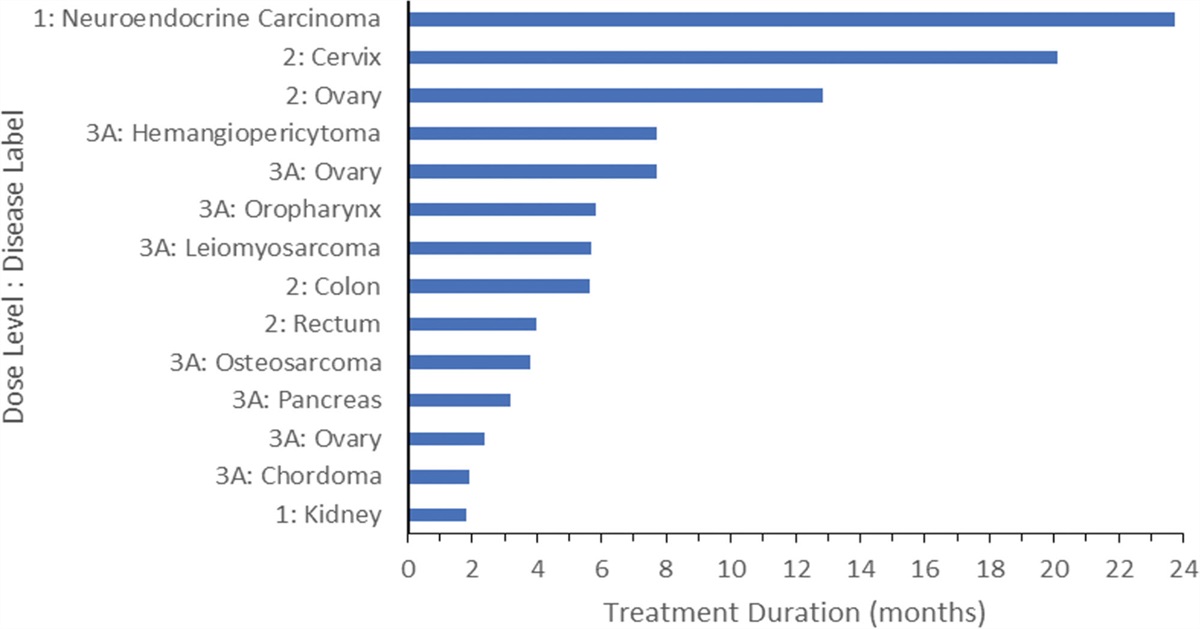
Disease responseFive patients did not meet the requirements for efficacy evaluability (3 patients stopped study treatment prior to Cycle 2 due to adverse events/disease complications, 1 patient withdrew from the study, and 1 patient died from causes unrelated to the study). Of the 24 patients who were evaluable for treatment efficacy, 14 patients (58%) had a best response of stable disease (SD) (Fig. 1). Ten patients experienced progressive disease. Patients with gynecologic malignancies had some of the longest durations of SD. One patient with progressive refractory neuroendocrine carcinoma tolerated treatment at DL1 for a total of 712 days and experienced SD throughout that time.
 Fig. 1:
Fig. 1: Treated patients with stable disease. Of the 24 patients who were evaluable for treatment efficacy, 14 patients showed a response of stable disease.
Correlative studies Pharmacodynamic studiesTo assess cytokine and growth factor levels in plasma, samples were collected at the same 5-time points used for the Multiplex Analysis and Pharmacokinetics study, as well as at baseline. Plasma samples from 23 patients were analyzed using The Human Immune Monitoring 65-plex ProcartaPlex Panel for MAGPIX (ThermoFisher) for the simultaneous detection and quantification of 65 protein targets. Of the studied proteins, there was a statistically significant reduction in HGF expression (F5, 73 = 3.15, p-value = 0.0126) (Fig. 2) between those who had SD for at least 4 weeks when compared to those who did not have SD for at least 4 weeks (total n = 23, n = 7 with SD). SDF1 levels appeared uniformly higher and CD30, and TNF RII levels appeared lower in patients who had SD for at least 4 weeks, but were not statistically significant. Archived tumor tissue samples from 25 patients were used to determine the level of PDE5 (Supplemental Table 1, Supplemental digital content 1, https://links.lww.com/ACD/A518), which showed no correlation to disease response.
 Fig. 2:
Fig. 2: Protein target analysis. Plasma samples from 23 patients were analyzed for detection and quantification of protein targets at 6 timepoints. (a) HGF showed a statistically significant reduction in expression between those who had SD for at least 4 weeks when compared to those who did not have SD for at least 4 weeks. (b) SDF1 levels appeared uniformly higher in patients who had SD for at least 4 weeks but were not statistically significant. (c) CD30 and (d) TNF RII levels appeared lower in patients who had SD for at least 4 weeks, but were not statistically significant.
Pharmacokinetic studiesBoth regorafenib and sildenafil are metabolized by the cytochrome p450 3A4 (CYP3A4) pathway. Therefore, PK analysis was performed to determine if the addition of sildenafil altered the PK profile of regorafenib. Blood samples were collected at the 6 timepoints listed above and processed as described in Materials & Methods and the plasma was analyzed by liquid chromatography equipped with tandem mass spectrometry. A total of 140 plasma samples from 31 patients were analyzed, (Supplemental Figure 1, Supplemental digital content 2, https://links.lww.com/ACD/A519). Analysis showed that sildenafil had no significant effect on the PK profile of regorafenib as seen from non-significant F-tests corresponding to the one-way ANOVAs comparing the three doses (1, 2 and 3A) at each of the time-points.
DiscussionOverall, the combination of regorafenib and sildenafil was well tolerated. Given the identified toxicities, the recommended phase 2 dose is regorafenib 160 mg with 100 mg of sildenafil daily. The toxicity profile of regorafenib and sildenafil was similar to FDA-approved monotherapy dosing [3]. Toxicity of regorafenib did not appear to be modulated by concurrent use of sildenafil.
While objective responses were not observed, 14 of 24 efficacy evaluable patients (58%) benefited from SD. The disease control in patients with gynecologic cancers was noteworthy, as 2 patients with ovarian cancer demonstrated SD for 7 and 12 months, respectively, and one patient with cervical cancer demonstrated SD for 20 months. The role of antiangiogenesis medications has been well-established for use in patients with ovarian malignancies. In first-line and recurrent settings, antiangiogenesis medications have been found to improve progression-free survival. They have also been found to improve overall prognosis by about 3 months in patients with high-risk disease. Given the established recommendations for use of antiangiogenic therapies in the adjuvant setting, the utilization of regorafenib with sildenafil could potentially be another modality of treatment based on the duration of SD observed patients with gynecologic malignancy in this study [26–31]. The SD responses of the patients with gynecologic cancers will be used as hypothesis-generating data for future studies.
In addition to the supported use in gynecologic malignancies, the use of VEGF inhibitors has been supported in patients with thyroid malignancies and neuroendocrine tumors [32,33]. Vandetanib has improved progression free survival in patients with unresectable, locally advanced or metastatic medullary thyroid cancer (30.5 vs. 19.3 months with placebo) though data on overall survival has not been established [34]. Extending the use of regorafenib in this setting may potentially add to treatment options for patients with advanced disease.
While no statistically significant alterations in the plasma levels of ligands for the EGF receptor family nor for cytokines such as IL6 and IL8 were found, we did observe a significant reduction in HGF expression in patients who exhibited prolonged SD. Hepatocyte growth factor, also known as scatter factor, promotes tumor cell growth, metastatic spread, and tumor angiogenesis. SDF1, also known as CXCL12, is a chemokine that promotes angiogenesis and inflammation and has been linked to tumor progression. CD30 is a member of the TNFR super-family and has elevated expression in T-cell and B-cell lymphomas. This data argues the drug combination is both suppressing growth and angiogenesis and metastatic spread of tumors. The combination of regorafenib and sildenafil appeared to reduce the levels of soluble TNFRII leading to reduction of T-regulatory cell activity. Given the known relationship between TNFRII and tumor immune evasion, drug combinations that reduce the level of TNFRII may allow increased efficacy of immunotherapy. In this phase 1 study, PDE5 staining is shown to be feasible and can be done in further phase 2 trials with more samples. However, due to the limited number of samples (with many from heavily pre-treated patients), a correlation of PDE5 staining to response cannot be determined in this study. Pharmacokinetic analysis also showed the feasibility of such an assay; however, due to the limited number of samples, a correlation to outcome cannot be determined.
We were also unable to correlate PDE5 expression to changes in plasma HGF levels. However, it is known that tumors express higher levels of PDE5 compared to matched normal tissues [35]. Sildenafil is a PDE5 inhibitor, which causes elevated cGMP levels and activation of PKG. One group has shown that PKG phosphorylates and inactivates c-MET, the HGF receptor [36]. It is tempting to speculate, therefore, that the actions of regorafenib plus sildenafil not only reduce HGF levels, but also act intracellularly to independently inactive c-MET signaling. Additional studies will be needed to answer this question.
Based on this data, the novel combination of regorafenib, sildenafil, and PDL1 inhibition could be of significant benefit among individuals with gynecologic malignancies.
LimitationsThe primary limitation of this study is the small study size. This study was not designed to evaluate for overall survival as the study population was heterogeneous. The patients enrolled in this study all had an ECOG performance status of 0 to 1, and many were heavily pre-treated with several lines of therapy. Given the inclusion criteria, the study population may not be representative of the population at large.
ConclusionThe combination of regorafenib and sildenafil at the recommended phase 2 dose is safe and generally well tolerated. Disease control in patients with gynecologic malignancies was especially encouraging. Further evaluation of the combination of regorafenib and sildenafil in gynecologic malignancies is warranted.
AcknowledgementsThis work used the Virginia Commonwealth University Massey Cancer Center Biostatistics Shared Resource and Tissue and Data Acquisition and Analysis Core. Regorafenib was provided by Bayer HealthCare Pharmaceuticals, Inc. Review and editing were performed by Molly R. Dickinson.
This work was supported by the National Cancer Institute of the National Institutes of Health (P30CA016059).
The study was reviewed and approved by the Institutional Review Board of Virginia Commonwealth University. The study was performed according to the principles defined by the Declaration of Helsinki.
Patient consent statement: All patients gave written informed consent prior to any study-related procedure.
Conflicts of interestThere are no conflicts of interest.
References 1. Mross K, Frost A, Steinbild S, Hedbom S, Büchert M, Fasol U, et al. A phase I dose-escalation study of regorafenib (BAY 73-4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res 2012; 18:2658–2667. 2. Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schütz G, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 2011; 129:245–255. 3. Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, et al.; CORRECT Study Group. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013; 381:303–312. 4. Demetri GD, Reichardt P, Kang YK, Blay J-Y, Rutkowski P, Gelderblom H, et al.; GRID study investigators. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013; 381:295–302. 5. Bruix J, Qin S, Merle P, Granito A, Huang Y-H, Bodoky G, et al.; RESORCE Investigators. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017; 389:56–66. 6. Goldstein I, Lue TF, Padma-Nathan H, Rosen RC, Steers WD, Wicker PA. Oral sildenafil in the treatment of erectile dysfunction. N Engl J Med 1998; 338:1397–1404. 7. Ghofrani HA, Wiedemann R, Rose F, Schermuly RT, Olschewski H, Weissmann N, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet 2002; 360:895–900. 8. Fisher DA, Smith JF, Pillar JS, St Denis SH, Cheng JB. Isolation and characterization of PDE9A, a novel human cGMP-specific phosphodiesterase. J Biol Chem 1998; 273:15559–15564. 9. Karami-Tehrani F, Moeinifard M, Aghaei M, Atri M. Evaluation of PDE5 and PDE9 expression in benign and malignant breast tumors. Arch Med Res 2012; 43:470–475. 10. Piazza GA, Speri G, Whitehead CM, Xu S, Kleinszanto A, Yipsneider M, et al. Cyclic GMP phosphodiesterase (cG PDE): Overexpression in human pancreatic carcinomas and a target for selective apoptotic antineoplastic drugs. Gastroenterology 2001; 120:A140. 11. Piazza GA, Klein-Szanto AJ, Xu S, Pamukcu R, Thompson WJ. Phosphodiesterase 5 overexpression in human non-small cell lung tumors compared to normal bronchial epithelium. Proc Am Assoc Cancer Res 2001; 42:811. 12. Piazza GA, Xu S, Klein-Szanto A, Ahnen DJ, Li H, Liu LI, et al. Overexpression of CGMP phosphodiesterase (cG PDE) in colonic neoplasias compared to normal mucosa. Gastroenterology 2000; 118:A282. 13. Wharton J, Strange JW, Møller GM, Growcott EJ, Ren X, Franklyn AP, et al. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am J Respir Crit Care Med 2005; 172:105–113. 14. Thompson WJ, Piazza GA, Li H, Liu L, Fetter J, Zhu B, et al. Exisulind induction of apoptosis involves guanosine 3’,5’-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated beta-catenin. Cancer Res 2000; 60:3338–3342. 15. Kumazoe M, Kim Y, Bae J, Takai M, Murata M, Suemasu Y, et al. Phosphodiesterase 5 inhibitor acts as a potent agent sensitizing acute myeloid leukemia cells to 67-kDa laminin receptor-dependent apoptosis. FEBS Lett 2013; 587:3052–3057. 16. Di X, Gennings C, Bear HD, Grah
留言 (0)