
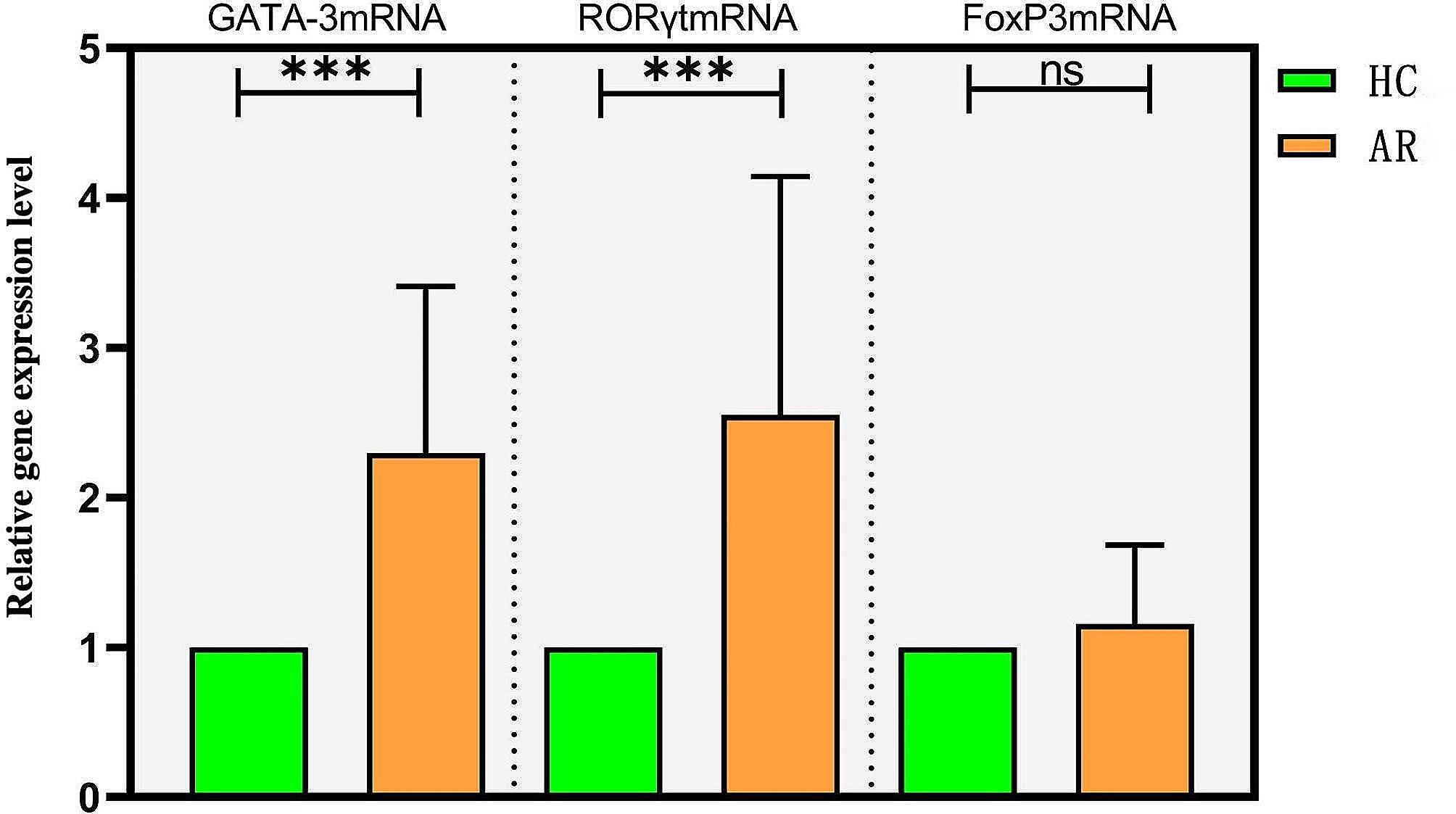
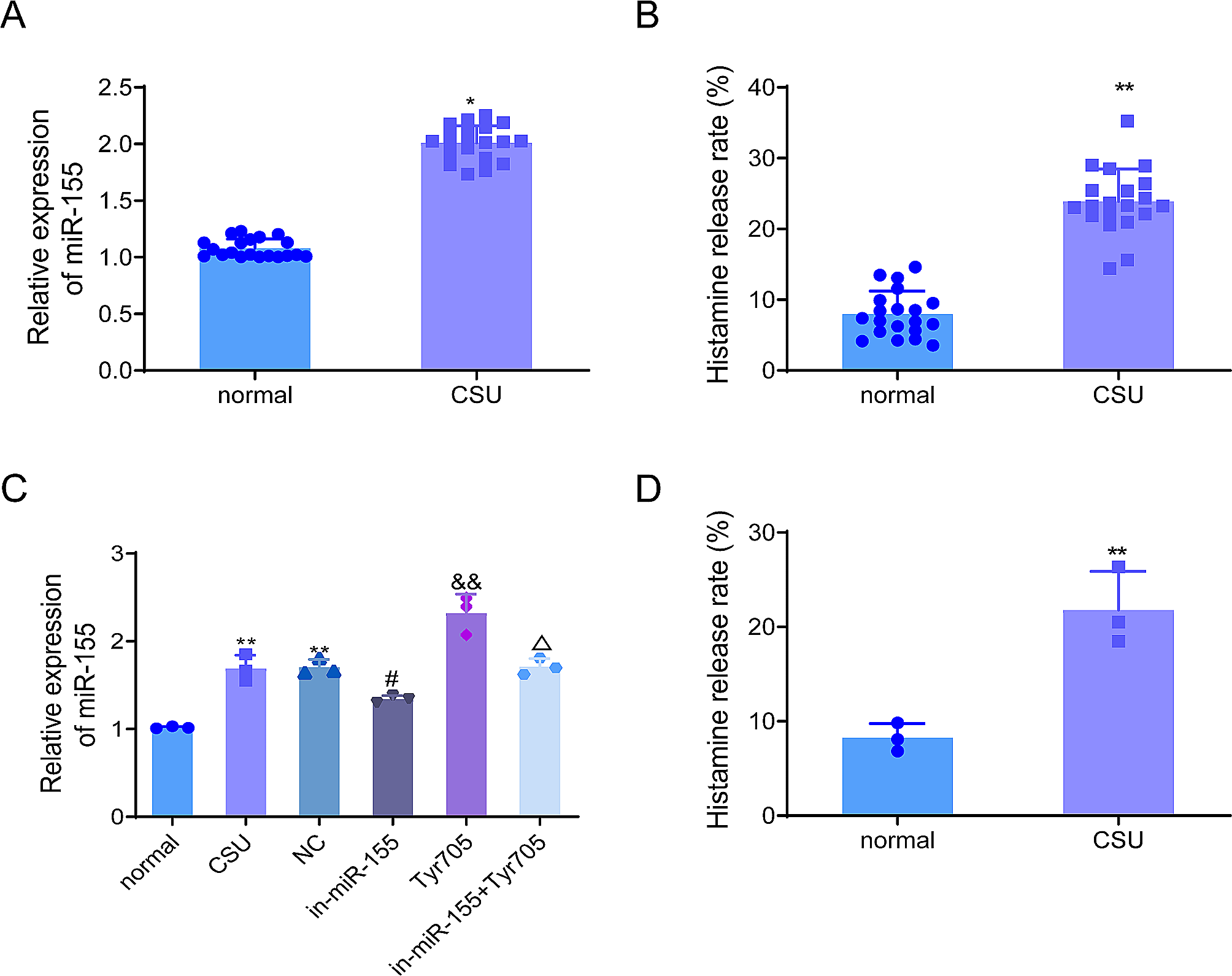
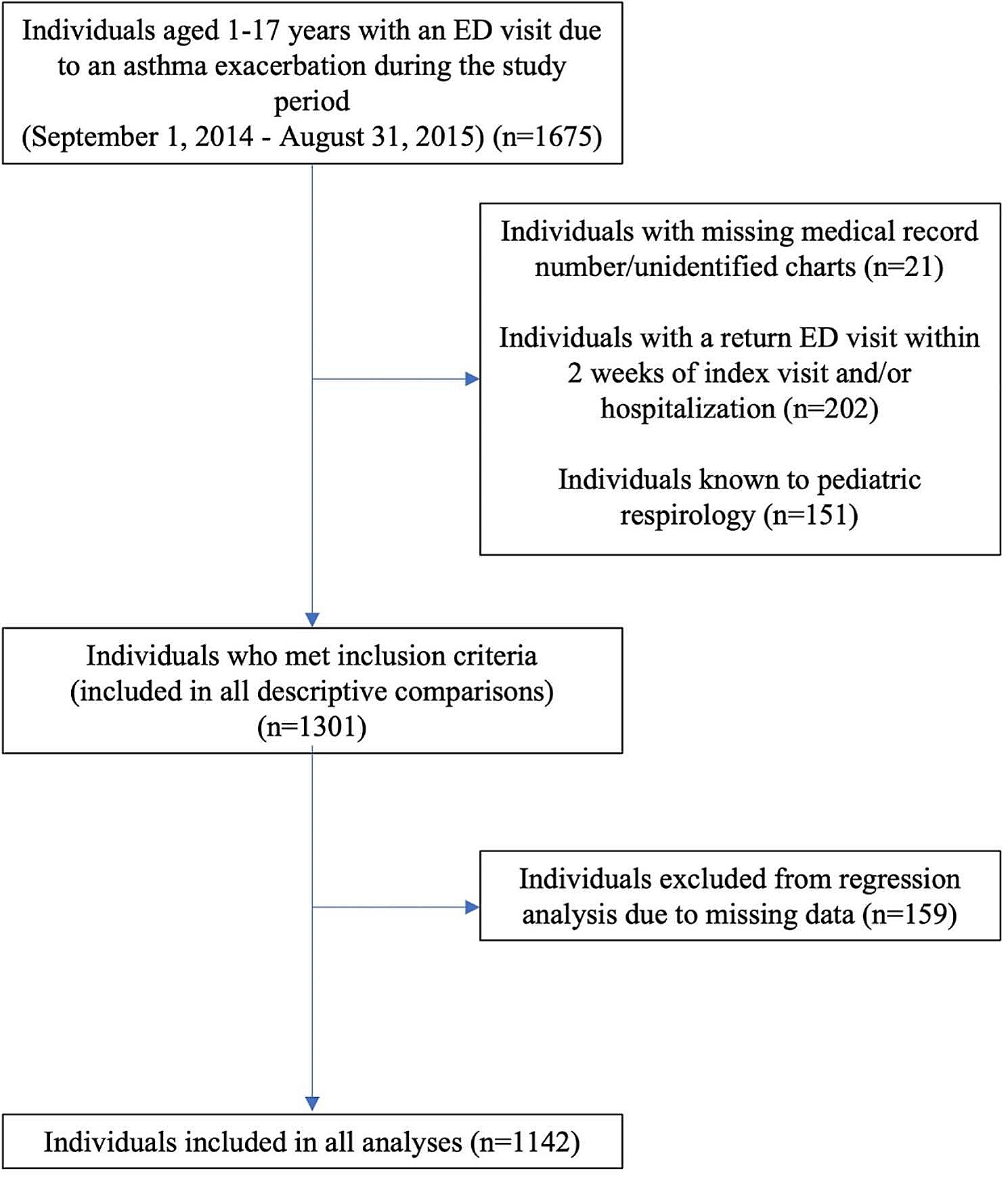
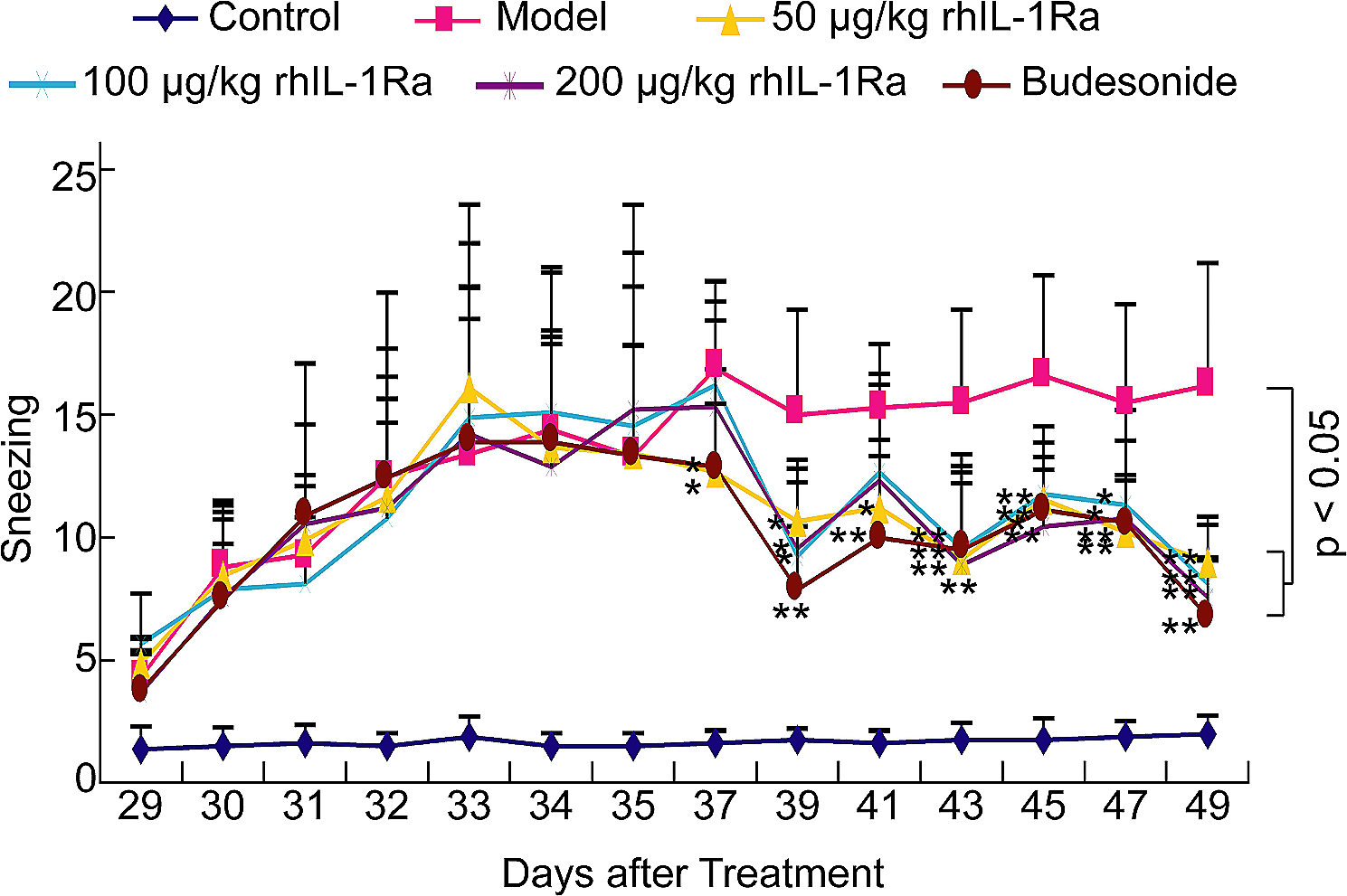
記住我



In our study, a cohort of 32 patients were enrolled, comprising nine patients presenting with clinical indications of HAE-I (both C1-INH and C1-INH activity were significantly low), one healthy relative of a proven HAE-I patient, one patient suspected of HAE-II (C1-INH activity was low with typical clinical signs), and 21 patients with suspected nC1-INH HAE. Employing conventional genetic testing methodologies (Sanger sequencing of SERPING1, PLG, and F12 genes) as described before [19], (likely) pathogenic genetic variants were successfully identified in eight (in seven out of nine investigated patients with diagnosed HAE-I and one out of 21 patients with nC1-INH-HAE) out of the 32 patients. Additionally, one patient with HAE-I had an intronic/splice region variant of uncertain significance in the SERPING1 gene. Patient 32, herein referred to as the relative of Patient 15, initially underwent testing as a healthy family member; however, the analysis revealed the presence of a pathogenic variant in the SERPING1 gene. Subsequent assessments demonstrated diminished levels of C1N and reduced C1 activity, leading to the asymptomatic diagnosis of HAE-I.
Genome sequencing provides definitive diagnosis and high confidence to re-evaluate clinical diagnosisInterestingly, the initial genetic testing failed to identify a pathogenic SERPING1 variant in three patients with suspected HAE-I or HAE-II (patient no. 8 had low levels of C1-INH ag, very low C1-INH activity, and low levels of C4; patient no. 22 also had low levels of C1-INH ag, low C1-INH activity, and low levels of C4; patient no. 11 had normal levels of C1-INH ag, but mildly decreased levels of C1-INH activity, and low C4). All tests were performed twice. Therefore, we performed genome sequencing for these three patients focusing first on coding SNVs (single nucleotide variants) and CNVs (copy number variations) in SERPING1, following by analysis of other SVs (structural variants) and non-coding SNVs. We were able to detect a pathogenic deletion of exon 4 with exact breakpoints at chr11[GRCh38]:g.57600729_57603011del (Figure S1) in one patient (Patient no. 22), confirming the diagnosis of HAE-I for this individual. The variant was not identified previously because MLPA (Multiplex ligation-dependent probe amplification) was not routinely applied for such cases.
In contrast, no pathogenic variants were found in the other two patients (patient no. 8 and 11) even after analysis of SVs and non-coding gene parts, prompting a re-evaluation of their diagnoses. After a two-year follow-up, in one of these patients (Patient no. 8) that had no family history of angioedema, paraproteins (monoclonal IgM) were eventually detected in the blood test of this patient, which were not present at the initial assessment two years prior. This led to confirming an alternative aetiology for the symptoms. Remarkably, the symptoms of another patient (Patient no. 11) with suspected HAE-II and no family history of angioedema resolved at follow-up, and the function of C1 activity returned to normal (C1-INH activity was mildly decreased twice), necessitating a revision of the diagnosis. These laboratory abnormalities could be related to the fact that the samples were shipped to another country for analysis, thus, longer shipping times. The patient is now considered to have bradykinin-related angioedema (antihistamine medications were found ineffective and other potential causes of angioedema were ruled out). Therefore, we show that genome sequencing can not only solve previously-unsolved cases, but also provide high confidence of an alternative cause for the “negative” cases.
Transcriptome sequencing provides intronic variant re-classificationFurthermore, among the patients diagnosed with HAE-I, one individual (Patient no. 9) was found to have a novel intronic splice region variant in the SERPING1 gene through Sanger sequencing NM_000062.2:c.1249 + 4 A > G, p?. The variant was predicted to result in a loss of canonical donor site (-4 bp) by Pangolin (splice loss = 0.71), but had low prediction confidence by spliceAI (donor loss = 0.16). Given the contradictory predictions and variant’s location in a non-canonical splice region, the actual effects were unknown. The segregation was limited to only one affected family member, therefore, without confirmation of a splicing defect, it was classified as a variant of uncertain significance. To evaluate the possible effects on splicing and assess the pathogenicity of this variant, whole transcriptome analysis with rRNA and globin depletion on peripheral blood-extracted RNA was performed. It revealed full intron retention on one allele in mRNA (with escaping nonsense mediated decay) (Fig. 1), successfully confirming the presence of a splicing defect due to the variant, allowing for the reclassification of the variant as pathogenic. This defect could easily be missed by targeted RT-PCR because the intron is ∼ 2.5Kb large and could fail the amplification.
Fig. 1
Retention of the last SERPING1 intron present due to the NM_000062.2:c.1249 + 4 A > G r.spl variant found in patient no. 9 using transcriptome sequencing. (A) cDNA sequence alignment to the genome in control (top) and patient (bottom) samples. Note increased coverage of a single allele of the last intron. (B) Sashimi plot of the splicing site usage in control (top) and patient (bottom) samples. Note low number reads covering the canonical splice site between the last two exons
Genetic testing of all currently known HAE (candidate) genes has low yield for nC1-INH-HAE, but is necessary for precise clinical diagnosisTo further investigate the genetic basis of nC1-INH-HAE in the remaining 20 patients, we performed exome sequencing, focusing on the analysis of coding SNVs and CNVs in recently-described nC1-INH-HAE (candidate) genes (F12, PLG, ANGPT1, KNG1, MYOF, and HS3ST6), as well other genes potentially involved in angioedema pathogenesis published previously [20] (total n = 55 genes; Table S1). However, no pathogenic or rare uncertain significance variants were detected in the genes of interest. Following these genetic analyses, all patients underwent a comprehensive re-evaluation of their clinical presentation, leading to a revision of their diagnoses.
Notably, none of the patients had a family history of angioedema attacks. As a result of the clinical re-evaluation after the negative result, 16 patients were ultimately diagnosed with bradykinin-related angioedema, while four patients were identified as having histaminergic angioedema since they reacted to treatment by Omalizumab.
In our study involving 32 patients with suspected HAE, encompassing various types and suspected nC1-INH HAE, conventional genetic testing identified pathogenic alterations in 10 patients. Genome sequencing for three unsolved patients revealed a previously-missed pathogenic variant, illustrating genome sequencing’s capacity to resolve challenging cases. Similarly Ren et al. [21]was able to solve all cases with HAE-I or HAE-II using genome sequencing for the initially-“negative” cases. Notably, the absence of pathogenic variants in two patients, despite extensive analysis, prompted a re-evaluation of their diagnoses; the emergence of paraproteins in one patient led to a revised diagnosis of histaminergic AE, while another patient’s symptoms resolved and their C1 activity normalised. Similarly, we were not able to identify a clinically-significant variant in 20 out of 21 nC1-INH HAE-suspected cases, even after exome sequencing with analysis of currently-confirmed and candidate HAE genes, which also resulted in a re-evaluation of their diagnosis (to different types of AE). This highlights the complex nature, symptom evolution, and variable clinical presentation of HAE and other angioedemas, and shows how even “negative” genetic results after comprehensive genetic testing can aid in reinterpretation of a clinical diagnosis. This dynamic nature of the disease challenges the static nature of genetic testing and highlights the importance of an integrative and continuous assessment of patient symptoms and genetic information.
Additionally, we highlight challenges in the interpretation of genetic results, particularly in the context of variants of uncertain significance. Functional validation of the variants to ascertain their clinical relevance is crucial, as exemplified by our reclassification of a novel SERPING1 splicing variant. Our findings show how application of transcriptome sequencing can help the reclassification of variants of uncertain significance that affect splicing.
Our findings indicate that the current diagnostic yield of nC1-INH HAE remains low. While newly proposed candidate genes have been reported as potentially involved in the pathogenesis of nC1-INH-HAE, there is no evident clinical justification for conducting genetic testing of these genes. Except for a single case with a pathogenic variant in the PLG gene, no pathogenic variants were identified among 20 patients in any of the described genes. Moreover, even if such variants were to be detected, they would likely be classified as variants of uncertain significance due to the limited described genotypic spectrum, providing limited assistance in the clinical diagnosis of the patients. Recently, another study sequenced 55 gene panels for 133 suspected-HAE individuals and failed to identify additional molecular diagnoses [20]. Further research is necessary to validate the association of these genes with nC1-INH HAE. Therefore, based on our and previous study results, sequencing of the described (candidate) genes has limited utility and should not be currently applied in diagnostic settings, except for F12 and PLG, which have a proven association with nC1-INH HAE. This experience underscores the complexity of the genetic landscape and the need for a more comprehensive understanding of the genetic factors contributing to nC1-INH HAE.
留言 (0)