
記住我



The PinC trial is an international, multicentre, randomised controlled superiority trial in infants born with CDH. Patients are currently recruited from nine tertiary care centres in the Netherlands, Belgium, Austria, Australia, Sweden, Italy, and Germany.
Study protocol development and conductThis trial has been approved by the Medical Ethical Committee of the Erasmus MC University Medical Center, Rotterdam, The Netherlands (MEC2019-0414). The trial is registered with the registry sponsored by the United States National Library of Medicine Clinicaltrials.gov NCT04373902 (registered April 2020). Local ethical approval was obtained from the ethical committees of participating centres before trial initiation. The study is conducted according to the principles of the Declaration of Helsinki, good clinical practice and international rules and regulations on personal data protection [10,11,12]. Changes in the trial protocol made after initiation of the trial are further explained in this paper.
Randomisation and data collectionInclusion criteria and exclusion criteria are reported in detail in the study protocol [9]. Infants are eligible if diagnosed with an isolated left-sided CDH on prenatal ultrasound with gestational age at delivery ≥ 35.0 weeks [9]. Exclusion criteria are right-sided and bilateral CDH, antenatal diagnosed major associated structural or genetic abnormalities, high urgency caesarean section (intended interval to delivery < 15 min), cases that have been treated during pregnancy with experimental drug therapy aiming to decrease the occurrence of pulmonary hypertension, twin pregnancies in which the infant diagnosed with CDH is born first, multiple birth > 2, and placental abnormalities (i.e. anterior placenta praevia, placental abruption). Written informed consent from both parents is required for inclusion. Eligible fetuses will be randomised 1:1 to either PBCC or the standard approach of immediate cord clamping. Study procedures regarding PBCC and immediate cord clamping are explained in the study protocol [9]. Blinding of the allocation arm during intervention is not possible due to the nature of the intervention. Allocation will be stratified by predicted lung size (determined by observed to expected lung-to-head ratio and liver position, graded as mild/moderate/severe lung hypoplasia, measured between 20 and 26 weeks of gestation or at initial visit) and by treatment centre, using variable random permutated block sizes (4-8) [13]. Randomisation and data collection are performed in the electronic data capture system Castor EDC. This electronic database facilitates on-site data entry and ensures concealment of allocation. Security is guaranteed with login names, login codes, and two factor authentication. Only dedicated and trained co-investigators in each centre receive credentials for Castor EDC and are thus allowed to enter data and randomise patients. Patient data are collected until the end of the study period, which is defined as discharge from the tertiary care hospital or death before discharge depending on which occurs first and with a maximum study duration of 6 months.
Data collection and management are implemented according to good clinical practice guidelines. To ensure feasibility of the trial, participating centres use their local CE-approved devices to assess the outcome measures strictly defined in the trial protocol. Those devices include amongst others resuscitation trolleys, respiratory function monitors, pulse oximeters, echocardiography machines, and laboratory equipment. All participating centres are certified tertiary academic hospitals that can carry out high-standard neonatal intensive care and all the trial-related investigational procedures. To increase reliability of the data, it is promoted to measure echocardiographic parameters in triplicate and averaged, although this is not possible in each centre. Biochemical and haematological outcomes, such as haemoglobin and bilirubin, are assessed in the local certified laboratories. Measurement units will be standardised as has been specified in Tables 1, 2, 3, and 4.
Table 1 Baseline characteristicsTable 2 Primary and secondary outcomesTable 3 Exploratory outcomesTable 4 Echocardiographic parameters [15]Baseline characteristicsBaseline characteristics will be collected for mothers and infants and will be presented in the final report of the trial. All collected data are depicted in Table 1.
Primary outcomeThe primary outcome is pulmonary hypertension diagnosed in the first 24 h after birth based on a combination of clinical and echocardiographic criteria, as was described in the study protocol [9]. Clinical parameters are as follows: (1) a difference between preductal and postductal oxygen saturation > 10% for a minimum of 15 min, with the specification of 15 min being added to the protocol after trial commencement because a single measurement of > 10% is likely due to a measurement error; (2) oxygenation index > 20. Echocardiographic parameters are as follows: (1) right ventricular systolic pressure ≥ 2/3 systemic systolic pressure; (2) right ventricle dilatation/septal displacement or right ventricular dysfunction ± left ventricular dysfunction [14, 15]. Pulmonary hypertension is present if at least 2 out of 4 criteria are present or if the infant requires ECMO therapy within the first 24 h after birth [14].
The initial version of the research protocol described echocardiographic evaluation between 12 to 24 h after birth. To guarantee feasibility in all centres, the trial team changed the evaluation period to within the first 24 h after birth, as routine evaluation in some centres takes places within the first 12 h. This change was reported to and approved by the medical ethical committee of the Erasmus MC in March 2020, before inclusion of the first patient. Furthermore, aiming to limit bias, the primary outcome was refined with the statement about ECMO therapy after discussions with additional centres in April 2021.
Secondary outcomes and exploratory outcomesTo limit type I errors, we predefined a limited number of secondary outcomes that will be included in formal statistical analyses. The choice of secondary outcomes was based on clinical relevance and existing literature and includes the following: (1) mortality before discharge from the tertiary care hospital, (2) presence of ≥ 3 criteria for pulmonary hypertension or extracorporeal membrane oxygenation within 24 h after birth, (3) ECMO therapy, (4) duration of supplemental oxygen need, (5) duration of mechanical ventilation, (6) duration of admission to the tertiary care hospital, and the safety parameter (7) postpartum haemorrhage (Table 2). All other secondary outcomes will be considered exploratory outcomes that will not be included in formal statistical testing. Additional to the exploratory outcomes depicted in Table 3, we will collect the following data: echocardiographic confirmation of the presence of pulmonary hypertension requiring therapy on days 7, 14, 21, and 28 and at discharge; the response to iNO defined as one of the following criteria: a decline of 10–20% in pre-postductal SpO2 difference, or an increase of 10–20% in PaO2, or improvement in haemodynamic parameters meaning 10% increase in mean blood pressure, or a decrease in lactate levels [6]. The number of days needing supplemental oxygen is defined as each calendar day on which the infant required FiO2 > 21% for any duration that day. For each calendar day of respiratory support, only the modality with the highest level of support applied on that day will be counted. Causes of death for deceased infants will be summarised in the final report. To objectify the echocardiographic criteria in the primary outcome, we will collect specific echocardiographic parameters in the first 24 h after birth, as depicted in Table 4 [15]. Where possible, the investigator evaluating the echocardiography is blinded to allocation, which will not be feasible in all centres due to limited human resources in routine practice. The first echocardiography including the parameters mentioned in Table 4 will be analysed.
Finally, continuous physiological measurements will be collected in the first 72 h if feasible, including the following: heart rate (bpm), preductal and postductal saturation (%), cerebral oxygenation (%), mean arterial blood pressure (mmHg), arterial partial pressure of oxygen (PaO2, kPa), and respiratory support settings (mean airway pressure in cmH2O, fraction of inspired oxygen in %, flow in L/min). Limited to infants born in the Erasmus MC, parental perception and appreciation of the approach during birth and stabilisation of their infant will be evaluated with a short questionnaire in both treatment arms. This questionnaire includes rating of 7 items on a 5-point scale and an open question. Topics include parental anxiety, safety, size of the team present, and provision of information. The results from physiological measurements and the parental questionnaire will not be analysed in the main report of the study but will be explored and reported separately.
SafetyAs CDH is a condition already associated with a significant risk of complications, serious morbidity is often inherent to the disease and unrelated to the intervention that is under evaluation in this trial. Therefore, we have specified context-specific SAEs that are reported to the data and safety monitoring board (DSMB) and METC on an annual base and that are collected as secondary outcomes: oxygen dependency on day 28, sepsis, cerebral complications, and ECMO therapy. Non-context specific SAEs are reported to the METC within 15 days after the sponsor has first knowledge of the SAE. Postpartum haemorrhage (estimated maternal blood loss > 1000 ml) is considered a safety parameter, and the sponsor will as such report this serious adverse event (SAE) to the METC within 7 days of first knowledge.
A DSMB was established to advise the principal investigator in protecting trial safety. Members of this committee are two neonatologists, an obstetrician, and a statistician. As stated in the protocol, the DSMB will conduct two interim statistical analyses on safety during the course of this study, after approximately 25% and 50% of the total required patients have completed their primary outcome. Outcomes included in the interim analysis on safety include the abovementioned context-specific SAEs, neonatal mortality, and postpartum haemorrhage. The first interim analysis has been conducted and resulted in the DSMB advising in favour of continuing the trial.
Statistical methods specified in the study protocolSample size calculationAs has been reported in the study protocol, the background incidence of pulmonary hypertension was previously reported at 69.7% in the first week after birth [9, 16]. Based on a suggested clinically relevant reduction in pulmonary hypertension incidence of one third, a total sample size of at least 140 infants was calculated with 80% power and a two-sided type I error of 5%.
Originally proposed analysesAfter start of the trial, the initially proposed analysis plan was updated to increase feasibility of the study in all participating centres and to limit bias where possible. Here, we present our updated statistical analysis plan.
Interim analyses and safety reportingAs was specified in the study protocol, no interim analyses on efficacy will be performed. Only two interim analyses on safety are planned, after 25% and 50% of the total required patients have completed the primary outcome. The only stopping condition will be concerns regarding safety outcomes. The decision to terminate or continue the trial is advised by the DSMB. The interim safety analyses include SAEs and the pre-specified context-specific safety outcomes listed as exploratory outcomes. Before each interim analysis, the DSMB will receive a report that includes blinded data on safety outcomes. On request of the DSMB, treatment allocation can be unblinded. The first safety interim analysis, after 25% of the total required patients had completed the primary outcome, was performed in 2022. The DSMB advised to continue the trial based on this interim analysis. In the final report of the trial, a list of SAEs and reasons of mortality will be reported by allocation arm in supplementary tables. SAEs and context-specific safety outcomes will be reported as is described in the ‘Statistical analyses’ section on secondary outcomes.
Statistical analysis planOverall principleStatistical significance is set at p < 0.05, using two-sided tests. For all relevant outcomes, a 95% confidence interval (CI) will be reported. Statistical analyses will be performed using the computing environment R (R Core Team (2020), Vienna, Austria).
Data handlingPotential outliers will be investigated, and extreme outliers, defined as being more than three times the interquartile (IQR) range below the first quartile or above the third quartile, will be listed individually in a supplement to the main analysis. If it can be reasonably assumed that those extreme outliers are due to an error in the data, they will be excluded from all analyses. The data set will only be locked after completion of the data set, data cleaning, and data validation. The statistical analyses for significant differences are done on a blinded data set and will be carried out after locking the data set, which can only be reversed in case of exceptional circumstances and after agreement of the trial team that consists of the principal investigator, data managers, and the trial statistician.
Definition of analysis setsThe primary outcome will be analysed in the intention-to-treat population to estimate the realised benefit of the intention to do PBCC over immediate cord clamping. The intention-to-treat population includes all patients that are randomised to a particular treatment arm (PBCC or immediate cord clamping), independent of actual treatment received, protocol deviations, or exclusion criteria. Patients will only be excluded from the study and thus from all analyses if parental consent is withdrawn. A secondary analysis will be performed in the per-protocol population to estimate the benefit of using PBCC—instead of having the intention to—over immediate cord clamping in the target population. The per-protocol population includes all randomised patients who completed the protocol for the arm they were assigned to, had the primary endpoint measured, had no major protocol violations, met all inclusion criteria, and did not meet any of the exclusion criteria. Relevant major protocol violations are limited to equipment-related decisions to deviate from the assigned protocol. For example, infants that are allocated to PBCC but receive immediate cord clamping due to the resuscitation trolley not being present will be analysed in the PBCC group in the intention-to-treat analysis but will be excluded from the per-protocol populations. Analysing these infants in the immediate cord clamping group could introduce bias, as we assume that cross-over will not be random. Additionally, we do not anticipate that infants receive PBCC despite being randomised for immediate cord clamping.
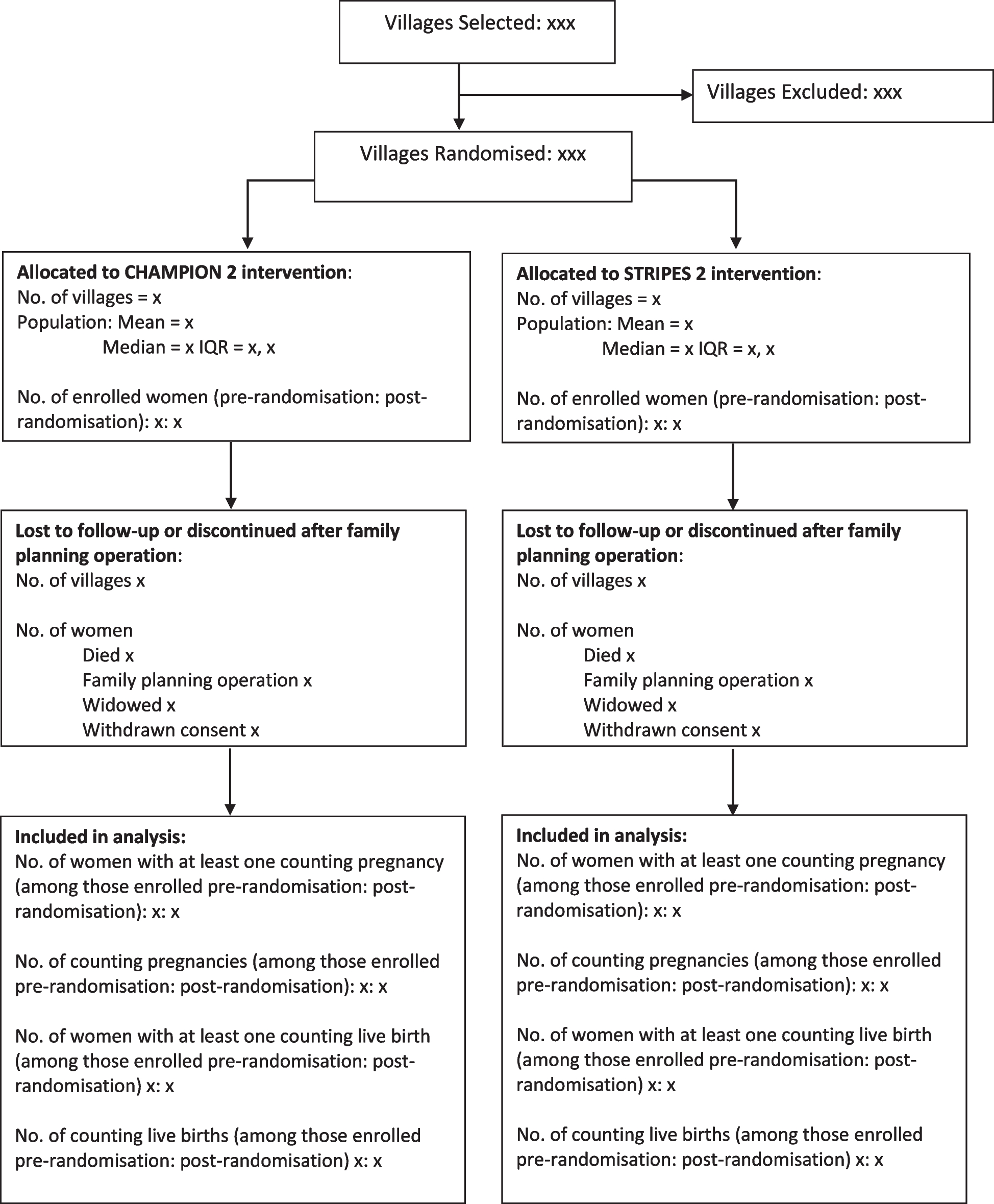
Statistical analysesPatient flowFigure 1 shows the expected patient flow. A similar figure, completed with numbers per category, will be included in the final report of the trial. We will summarise the reasons why patients are not eligible and the reasons for not including eligible patients if reported. Protocol deviations, defined as deviations in eligibility criteria and patients not being stabilised according to the allocated protocol as set forth in the study protocol, will also be reported.
Fig. 1
Flowchart of inclusion. CDH, congenital diaphragmatic hernia; ICC, immediate cord clamping; PBCC, physiological-based cord clamping
Baseline characteristicsAll baseline characteristics will be described for each allocation arm of the trial as depicted in Table 1. No formal statistical testing will be performed on baseline characteristics. Continuous data will be reported as mean ± SD or median [IQR] depending on distribution, and categorical data will be reported as counts and percentages.
Primary outcomeThe effect of PBCC on the primary outcome will be analysed as a complete case analysis in the intention-to-treat population. The main effect of PBCC on pulmonary hypertension will be analysed using the chi-square test. To explore the effect of PBCC on pulmonary hypertension per strata, subgroup analysis per strata (estimated mild/moderate/severe pulmonary hypoplasia) will be performed. An additional sensitivity analysis applying mixed effect models will be conducted to investigate the specific strata. The primary outcome will also be reported as absolute differences in percentages with 95% CI and relative risks with 95% CI to compare the intervention group with the control group (Table 2).
Analysis of the primary outcome will be based on complete cases and by protocol the independent variables in this multivariable analysis cannot be missing as these are required for randomisation. In the rare event that evaluation of the primary outcome has not been performed in the first 24 h, e.g. in the event of mortality before echocardiographic evaluation, the dependent outcome (i.e. pulmonary hypertension) will be missing. To evaluate the robustness of our findings and potential bias by such missing data, we will perform a sensitivity analysis. In this sensitivity analysis, missing values will be imputed by using the ‘worst case’ observed in cases in which the primary outcome was assessed.
Secondary outcomesTo limit multiplicity, formal statistical analyses will be carried out for only a limited number of pre-defined secondary outcomes that were regarded most relevant from a clinical perspective (Table 2). When mortality competes with outcomes, the risk for these outcomes can be underestimated. Therefore, these secondary outcomes will be reported in both the group of survivors and the total study population. For continuous variables, we will report absolute differences in means and medians with 95% CI. For dichotomous variables, we will report relative risks with 95% CI and absolute differences in percentages with 95% CI. The results will not be adjusted for multiplicity and p-values will not be calculated. Additionally, the distribution of the data for each of the pre-defined continuous secondary outcomes will be presented in histograms.
All possible effort will be made to complete the dataset and we expect that data on the predefined secondary outcomes will be present in nearly all infants. Hence, we will not use imputation to complete the dataset in case of missing values. The number of missing data in secondary outcomes will be reported.
Exploratory secondary outcomesDescriptive statistics will be used to report all exploratory secondary outcomes—including context-specific safety outcomes—in the intervention group and control group, separately, and formal statistical testing will thus not be performed. Continuous data will be reported as mean ± SD or median [IQR] depending on distribution, and categorical data will be reported as counts and percentages.
Trial reporting and statusThe trial will be reported following the principles laid out in the CONSORT statement [17]. The trial was initiated in the Erasmus MC in May 2020. Eight additional centres started recruitment of patients between August 2020 and March 2024. We anticipate on including the final patient end-2024.
留言 (0)