
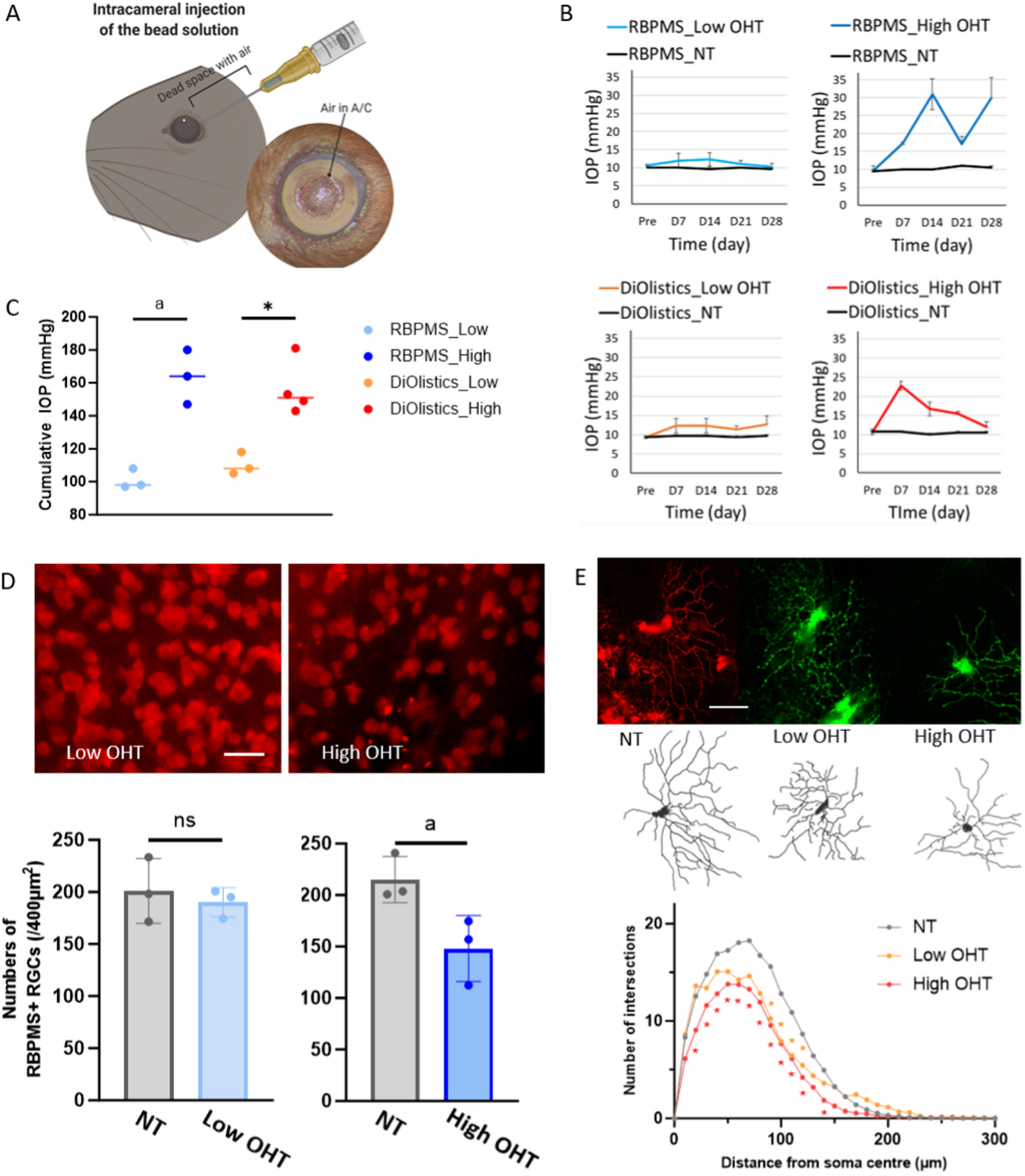
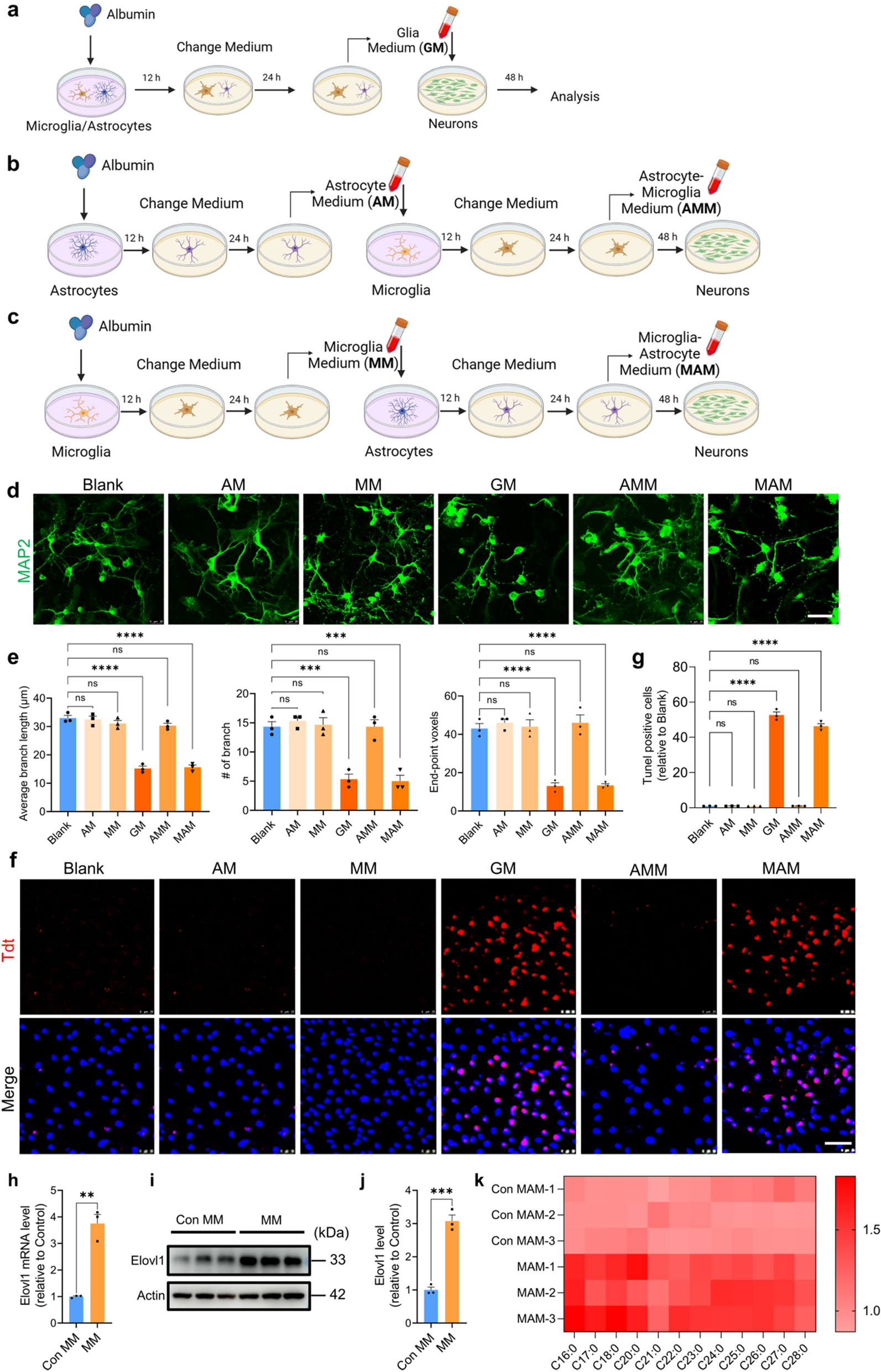
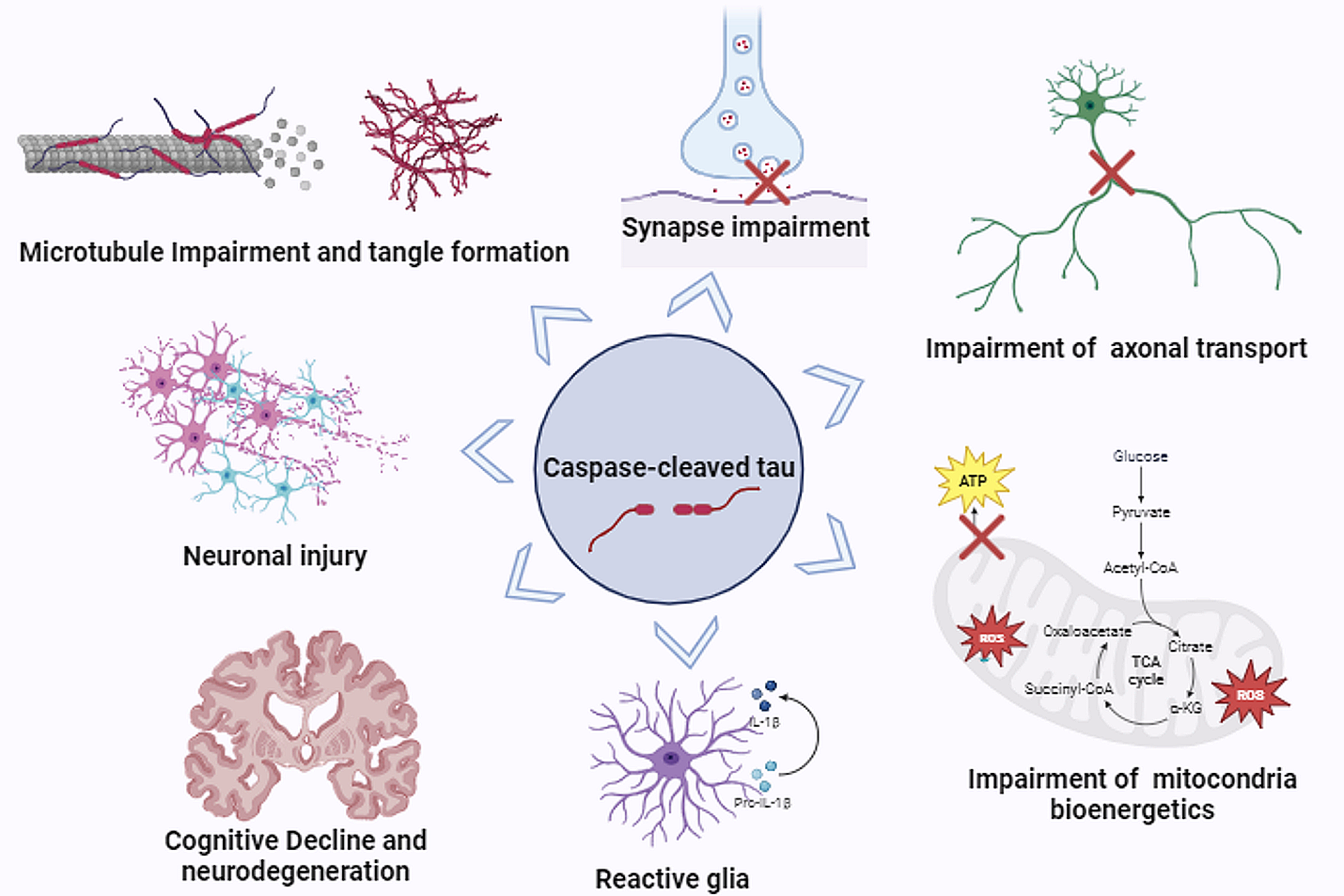
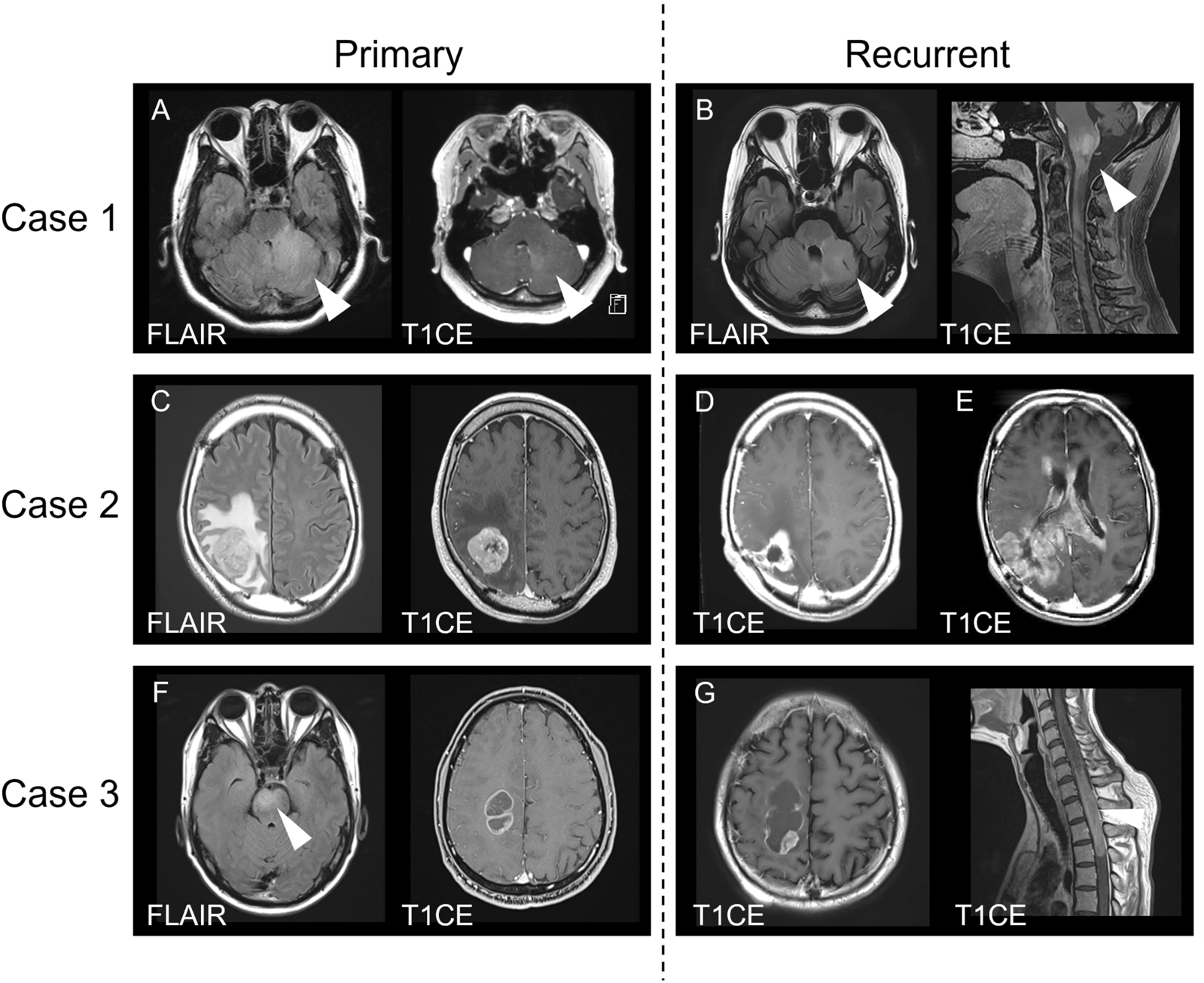
記住我





To evaluate how inhibition of β1 integrin function impacts vascular integrity of spinal cord blood vessels, young (8–10 weeks) mice were exposed to chronic mild hypoxia (CMH, 8% O2) or normoxic control conditions for 4 days, and received either daily intraperitoneal (i.p.) injections of the anti-mouse β1 integrin function-blocking antibody HMβ1-1, or an isotype control antibody (at doses of 2.5 mg/kg) for the duration of the 4 day CMH treatment. To confirm that the hamster anti-mouse β1 integrin blocking antibody localizes to blood vessels in spinal cord tissue, we conducted immunofluorescent (IF) studies with an anti-hamster secondary antibody. This demonstrated that the hamster blocking antibody localized very specifically to blood vessels (Supplementary Fig. 1). Vascular leak was evaluated by dual-IF using CD31 to identify endothelial cells and fibrinogen to detect extravascular leak. As shown in Fig. 1A, under normoxic conditions, while no spinal cord vascular leak was detected in mice receiving isotype control antibody, β1 integrin blockade triggered a small number of vascular leaks. However, under hypoxic conditions, while a relatively small number of leaks occurred in control antibody treated mice, β1 integrin inhibition greatly enhanced both the total number (\( \sim \)22-fold) and area (\( \sim \)65-fold) of spinal cord vascular leaks (Fig. 1A-C). Strikingly, most of these leaks occurred in the WM (shown in Fig. 1A and quantified in Fig. 1C). Dual-IF with fibrinogen and the WM marker fluoromyelin confirmed that almost all the hypoxia-induced vascular leaks occurred in the myelinated WM region of spinal cord (Fig. 1D). Of note, areas of vascular leak were strongly associated with marked loss of fluoromyelin signal, implying that vascular disruption erodes myelin integrity. Furthermore, triple-IF with CD31, fibrinogen, and the erythrocyte marker TER-119 demonstrated that many of these vascular disruptions were severe enough to result in hemmorhage into surrounding tissue, as shown by the extravascular leak of TER-119+ erythrocytes. In line with the fibrinogen data, most of these hemorrhages were also restricted to WM (Fig. 1E). From these findings we conclude first, that β1 integrins play an essential role in the maintenance of vascular integrity in spinal cord blood vessels, second, that this function is particularly critical during hypoxia-induced vascular remodeling, and third, this function appears to be especially relevant to WM blood vessels. To examine how β1 integrin inhibition impacts endothelial proliferation in spinal cord blood vessels, we performed dual-IF with CD31 and the proliferation marker Ki67 in the same group of mice. Consistent with our previous findings [17], this revealed that CMH strongly promoted endothelial proliferation in the spinal cord, with WM showing the strongest response (Supplementary Fig. 2B). Of note, β1 integrin blockade had no obvious effect on the rate of endothelial proliferation in the hypoxic spinal cord, either in WM or GM (Supplementary Fig. 2).
Fig. 1
β1 integrin inhibition greatly increases hypoxia-induced spinal cord vascular disruption, preferentially in WM. Frozen spinal cord sections taken from mice exposed to normoxia or hypoxia (8% O2) that had received daily intraperitoneal injections of the anti-mouse β1 integrin function-blocking antibody or isotype control antibody for 4 days were stained for CD31 (AlexaFluor-488) and fibrinogen (Cy-3) (A), fluoromyelin (red) and fibrinogen (AlexaFluor-488) (D), or CD31 (AlexaFluor-488), fibrinogen (Cy-5), or TER-119 (Cy-3) (E). Scale bars = 500 μm, except for high power (HP) images in lower panel of A, where scale bar = 200 μm. White dotted line demarcates the GM (inside) from the WM (outside). (B-C) Quantification of the total area (B) or number of vascular leaks/FOV (C) in the spinal cord after normoxia or 4-days hypoxia. Results are expressed as the mean ± SEM (n = 6–10 mice/group). *** p < 0.001. Note that β1 integrin inhibition greatly increased the extent of hypoxia-induced vascular disruption in the spinal cord, preferentially in the WM
β1 integrin inhibition greatly enhances hypoxia-induced microglial activationAs we have shown that hypoxia-induced CNS vascular disruption results in a marked microglial activation response [13,14,15], we next investigated how β1 integrin inhibition impacts microglial activation in the spinal cord by performing dual-IF with Mac-1/fibrinogen and CD68/fibrinogen. This showed that under normoxic conditions, Mac-1+ microglia occupied a ramified inactivated morphology (Fig. 2A), along with low levels of CD68 (a marker of microglial priming; Fig. 2C). Under normoxic conditions, β1 integrin inhibition induced a very small, but non-significant increase in these markers. Under hypoxic conditions, the isotype control antibody also triggered a small non-significant upwards trend of microglial activation markers compared to the normoxic baseline, but most strikingly, β1 integrin inhibition triggered a very strong microglial activation response in both the WM and GM compartments of spinal cord as shown by marked morphological transformation into the classical activated phenotype (large cell body with short processes) and increased number and size of CD68+ cells (Fig. 2A-B). As microglial proliferation is an integral part of a strong microgliosis response, we also evaluated this by Mac-1/Ki67 dual-IF (supplementary Fig. 3). This showed that the density of proliferating microglia was very low under normoxic conditions. After hypoxic exposure, while proliferating microglia were rarely present under isotype control antibody conditions, this number was strongly increased by β1 integrin inhibition, indicating a strong microglial proliferation response (see arrows in Supplementary Fig. 3A). Notably, enhancement of microglial activation was strongest in the WM (Fig. 2E-F and Supplementary Fig. 3B), concordant with the greater levels of vascular disruption in this region.
Fig. 2
β1 integrin inhibition greatly enhances microglial activation in the hypoxic spinal cord. Frozen spinal cord sections taken from mice exposed to normoxia or hypoxia (8% O2) that had received daily intraperitoneal injections of the anti-mouse β1 integrin function-blocking antibody or isotype control antibody for 4 days were stained for Mac-1 (AlexaFluor-488) and fibrinogen (Cy-3) (A) or CD68 (AlexaFluor-488) and fibrinogen (Cy-3) (C). Lower magnification images of Mac-1 and CD68 IF are shown in B and D, respectively. Scale bars = 50 μm (A and C) or 200 μm (B and D). White dotted line demarcates the GM (inside) from the WM (outside). Quantification of the number of morphologically activated microglia/FOV (E) or number of CD68 + microglia/FOV (F) after 0- or 4-days hypoxia. Results are expressed as the mean ± SEM (n = 6 mice/group). *** p < 0.001. Note that β1 integrin inhibition strongly increased all parameters of microglial activation in the hypoxic spinal cord
β1 integrin inhibition triggers loss of oligodendroglial cells specifically in WMBecause fluoromyelin/fibrinogen dual-IF indicated a strong correlation between vascular leak and myelin degradation (Fig. 1D), we next evaluated the impact of β1 integrin inhibition on cells of the oligodendroglial lineage. First, we quantified by IF the cell density of mature oligodendrocytes using the CC1 marker (Fig. 3A). This revealed that hypoxia alone had no noticeable impact on the density of CC1 + cells, and that under normoxic conditions, β1 integrin inhibition had no discernible influence on CC1 + cell density. However, in the presence of hypoxia, β1 integrin inhibition significantly reduced the density of CC1 + cells specifically in the WM (Fig. 3A-B). We next examined how β1 integrin inhibition influences the density of oligodendrocyte precursor cells (OPCs) by staining for platelet derived growth factor receptor alpha (PDGFRα). In a similar manner to CC1, PDGFRα IF revealed that hypoxia alone did not affect the density of OPCs, and that under normoxic conditions, β1 integrin inhibition had no discernible influence on OPC density, but in the presence of hypoxia, β1 integrin inhibition significantly reduced the density of OPCs specifically in the WM (Fig. 3C). These data are consistent with our finding that under hypoxic conditions, β1 integrin inhibition triggers greatest vascular disruption in WM (Fig. 1), which in turn leads to greater loss of oligodendroglial cells in this region. Dual-IF with the erythrocyte marker TER-119 and the oligodendrocyte marker CC1 showed that in WM regions showing large vascular leaks, extravascular deposition of erythrocytes correlated with striking loss of oligodendrocytes (note the two dark holes corresponding to the TER-119+ region in the lower middle panel of Fig. 3D).
Fig. 3
β1 integrin inhibition triggers loss of oligodendroglial cells specifically in WM. Frozen spinal cord sections taken from mice exposed to normoxia or hypoxia (8% O2) that had received daily intraperitoneal injections of the anti-mouse β1 integrin function-blocking antibody or isotype control antibody for 4 days were stained for CC1 (top two rows)) or PDGFRα (lower two rows) (A) or TER-119 (AlexaFluor-488) and CC1 (Cy-3) (D). Scale bars = 50 μm (A) or 100 μm (D). Quantification of the number of CC1 + cells/FOV (B) or PDGFRα + cells/FOV (C) after 0- or 4-days hypoxia. Results are expressed as the mean ± SEM (n = 6 mice/group). * p < 0.05. Note that β1 integrin inhibition decreased the cell density of mature oligodendrocytes and OPCs in the hypoxic spinal cord, specifically in the WM. Also note that vascular disruption led to obvious loss of oligodendrocytes in the immediate vicinity of erythrocyte deposition (D)
In conditions of chronic mild hypoxia, spinal cord tissue hypoxia is most severe in the WMOur observation that under hypoxic conditions, β1 integrin inhibition disrupts vascular integrity preferentially in WM demonstrates a novel and fundamental difference between WM and GM blood vessels. This raises the question: why are WM blood vessels so vulnerable to disruption while GM vessels are relatively resistant? One possibility is that the expression of molecules contributing to vascular integrity differs between WM and GM. For instance, if WM blood vessels express lower levels of β1 integrins, one might expect the blocking antibody would more effectively block β1 integrin function in WM before it does in GM. To address this possibility, we compared WM and GM for vascular expression of cell adhesion receptors (the integrin subunits β1, α5 and α6, and dystroglycan, Supplementary Fig. 4A), the ECM proteins fibronectin, laminin, collagen IV and perlecan (Supplementary Fig. 4B), tight junction proteins claudin-5, occludin, and ZO-1 and the junctional adhesion protein VE-cadherin (Supplementary Fig. 4C), and markers of pericyte coverage (CD13 and PDGFRβ, Supplementary Fig. 4D). This confirmed first that vascular density in GM is \( \sim \)22-fold) and area (\( \sim \)4 times higher than in WM, and second, that vascular expression of almost all molecules examined was noticeably higher in hypoxia treated mice both in the WM and GM regions. However, at the single vessel level, we did not observe any major WM/GM differences in the expression levels of any of these important BBB molecules.
An alternative possibility that might account for the greater vulnerability of WM vessels to hypoxia is that differences in vascular density or architecture are responsible. Specifically, vessel density in GM is 4 times greater than WM; thus, the average vessel to cell diffusion distance is 4 times greater in WM [17, 19, 34]. In keeping with this greater vascular density, cerebral blood flow has been shown to be 4 times greater in GM compared to WM [10, 24]. Based on these observations, it has been suggested that GM tissue has a much greater vascular reserve than WM [10, 24]. Thus, when oxygen is limited, WM tissue is far more likely to experience greater levels of hypoxia, and as hypoxia triggers angiogenesis and vascular breakdown, the result is more vascular disruption in WM. To directly test this idea in the CMH model, we evaluated the appearance of hypoxia in the spinal cord by i.p. administration of hypoxyprobe (pimonidazole hydrochloride), which forms permanent protein adducts in hypoxic cells when oxygen levels dip below 1.5% oxygen (pO2 < 10 mmHg), and which can subsequently be detected by polyclonal antibody [1, 32]. As expected, the normoxic spinal cord contained no hypoxyprobe+ regions or vascular leaks. In contrast, spinal cord sections taken from CMH mice contained many hypoxyprobe+ regions and importantly, the vast majority of these were in the WM (Fig. 4A-B). Interestingly, while β1 integrin inhibition did not change the density or WM/GM distribution of hypoxyprobe+ regions, it triggered a strong increase in the number of vascular leaks (Fig. 4A) and most of these were in the WM (Fig. 4B). Dual-IF with the erythrocyte marker TER-119 and hypoxyprobe confirmed that the majority of hypoxyprobe+ regions occur in the WM, and that hypoxia, superimposed with β1 integrin blockade, results in greatly increased numbers of vascular leaks, specifically in spinal cord WM (Fig. 4C-D). Together, these data show that in mice exposed to CMH, spinal cord WM is far more susceptible to manifesting regions of hypoxia and vascular disruption. Taken with our previous observation that WM launches a more vigorous angiogenic response to hypoxia than GM [17], we propose a model that explains all these findings: (i) the low vessel density (and blood flow) in WM predisposes to greater susceptibility to hypoxia, (ii) this hypoxia drives angiogenesis at a faster rate in WM, and (iii) the greater extent of angiogenic remodelling in WM, which involves uncoupling of endothelial cells from ECM proteins, allows the β1 blocking antibody greater access to interfere with endothelial-ECM interactions, resulting in higher levels of vascular disruption in WM.
Fig. 4
In the chronic mild hypoxia model, tissue hypoxia is most severe in the WM. Frozen spinal cord sections taken from mice exposed to hypoxia (8% O2) that had received daily intraperitoneal injections of the anti-mouse β1 integrin function-blocking antibody or isotype control antibody for 4 days, followed by an intraperitoneal injection of hypoxyprobe 2 h before tissue samples were collected, were stained for fibrinogen (Cy-3) and hypoxyprobe (AlexaFluor-488) (A), or TER-119 (AlexaFluor-488) and hypoxyprobe (Cy3) (C). Scale bars = 500 μm. White dotted line demarcates the GM (inside) from the WM (outside). B. Quantification of the number of hypoxyprobe + events/FOV in the spinal cord after 4-days hypoxia. Results are expressed as the mean ± SEM (n = 4–5 mice/group). Note that most hypoxyprobe + events occurred in the WM, that many vascular leaks, but not all, were associated with a hypoxyprobe + region, and that β1 integrin inhibition while greatly increasing the number of vascular leaks, had no effect on the appearance or distribution of hypoxic regions
White matter tracts in the brain also show greater susceptibility to β1 integrin inhibition-induced vascular leak under hypoxic conditionsBased on our spinal cord findings that β1 integrin inhibition greatly increases hypoxia-induced vascular disruption almost exclusively in WM, we next extended our analysis to the brain to see if our proposed model also holds true there. Here we examined two well demarcated WM tracts; the WM tracts of the cerebellum and the corpus callosum, and compared them with the surrounding GM areas of the cerebellum and cerebral cortex, respectively. First, we compared the angiogenic response in WM and GM regions by quantifying the density of proliferating endothelial cells, using CD31/Ki67 dual-IF. This showed that in the cerebellum, almost all the proliferating endothelial cells were in the WM tract, with very few in the neighboring GM (Fig. 5A). This was confirmed using fluoromyelin/Ki67 dual-IF (Fig. 5B). Quantification of CD31+/Ki67+ cells in the cerebellum revealed a WM:GM ratio of 43:1 (Fig. 5C). In a similar manner, in the forebrain, the WM corpus callosum tract contained a much higher density of proliferating endothelial cells than the neighboring cerebral cortex (Fig. 5D), which was confirmed by fluoromyelin/Ki67 dual-IF (Fig. 5E). Quantification of CD31+/Ki67+ cells in the WM corpus callosum and the GM cerebral cortex revealed a WM:GM ratio of 5:1, (Fig. 5F). Thus, consistent with our observations in spinal cord WM [17], in both these brain regions, the WM contained a much greater number of proliferating endothelial cells. Next, we compared the relative distribution of BBB disruption in the WM and GM regions of the cerebellum and the forebrain (cerebral cortex and corpus callosum). Similar to spinal cord, this revealed that under normoxic conditions, no BBB disruption was detected in mice receiving isotype control antibody, while β1 integrin blockade triggered a very small number of vascular leaks (Fig. 6). However, under hypoxic conditions, while a small number of leaks occurred in control antibody treated mice, β1 integrin inhibition greatly enhanced the number of vascular leaks both in the cerebellum and the forebrain (\( \sim \)75-fold and 13-fold respectively compared to isotype control conditions). Most strikingly, these vascular leaks occurred almost exclusively in the WM, both in the cerebellum (Fig. 6A) and the corpus callosum of the forebrain (Fig. 6C). In the cerebellum, almost all vascular leaks occurred in the WM tract, with very few in the surrounding GM (WM:GM ratio of 41:1, Fig. 6E), while the corpus callosum WM tract contained many vascular leaks with just a few in the surrounding cerebral cortex GM (WM:GM ratio of 12, Fig. 6F). Fluoromyelin/fibrinogen dual-IF confirmed that almost all vascular leaks identified in these brain regions occurred in the myelinated WM regions (Fig. 6B and D). In addition, in both the cerebellum and forebrain, fluoromyelin/fibrinogen dual-IF demonstrated a strong spatial association between vascular disruption and myelin degradation like that found in the spinal cord (note the motheaten appearance of myelin in Fig. 6B and D). These observations support the concept that the preferential tendency for WM (in both brain and spinal cord) to show vascular disruption in response to CMH in the presence of β1 integrin blockade, is a result of lower vascularity triggering stronger angiogenic remodelling, allowing the β1 integrin blocking antibody more opportunity to prevent the stabilization of newly formed blood vessels in these regions.
Fig. 5
Hypoxia-induced endothelial proliferation in the brain occurs preferentially in white matter tracts. Frozen brain sections taken from mice exposed to normoxia or hypoxia (8% O2) for 4 days were stained for CD31 (AlexaFluor-488), Ki67 (Cy-3), and DAPI (blue) (A, cerebellum and D, forebrain) or fluoromyelin (red) and Ki67 (AlexaFluor-488) (B, cerebellum and E, forebrain). Scale bars = 500 μm except for high power (HP) images where scale bars = 200 μm. White dotted line demarcates the WM corpus callosum (CC, inside) from the surrounding cerebral cortex (CX) grey matter. C and F. Quantification of the number of proliferating endothelial cells/unit area in the cerebellum (C) or cerebral cortex/corpus callosum areas (F) after 0- or 4-days hypoxia. Results are expressed as the mean ± SEM (n = 5–6 mice/group). *** p < 0.001. Note that in both brain areas, most of the hypoxia-induced endothelial proliferation occurs in the WM tracts
Fig. 6
White matter tracts in the brain show a similar predilection to β1 integrin inhibition-induced vascular leak under hypoxic conditions. Frozen brain sections taken from mice exposed to normoxia or hypoxia (8% O2) that had received daily intraperitoneal injections of the anti-mouse β1 integrin function-blocking antibody or isotype control antibody for 4 days were stained for CD31 (AlexaFluor-488), fibrinogen (Cy-3) and DAPI (blue) (A and C) or fluoromyelin (red) and fibrinogen (AlexaFluor-488) (B and D). A-B and C-D images show the cerebellum and cerebral cortex/corpus callosum, respectively. Scale bars = 500 μm. White dotted line demarcates the WM corpus callosum (inside) from the surrounding grey matter. E-F. Quantification of the number of vascular leaks/unit area in the cerebellum (E) or cerebral cortex/corpus callosum areas after 0- or 4-days hypoxia. Results are expressed as the mean ± SEM (n = 6–10 mice/group). *** p < 0.001. Note that under hypoxic conditions, β1 integrin inhibition greatly increased the extent of vascular disruption in the brain, preferentially in the cerebellar and corpus callosum WM tracts
留言 (0)