
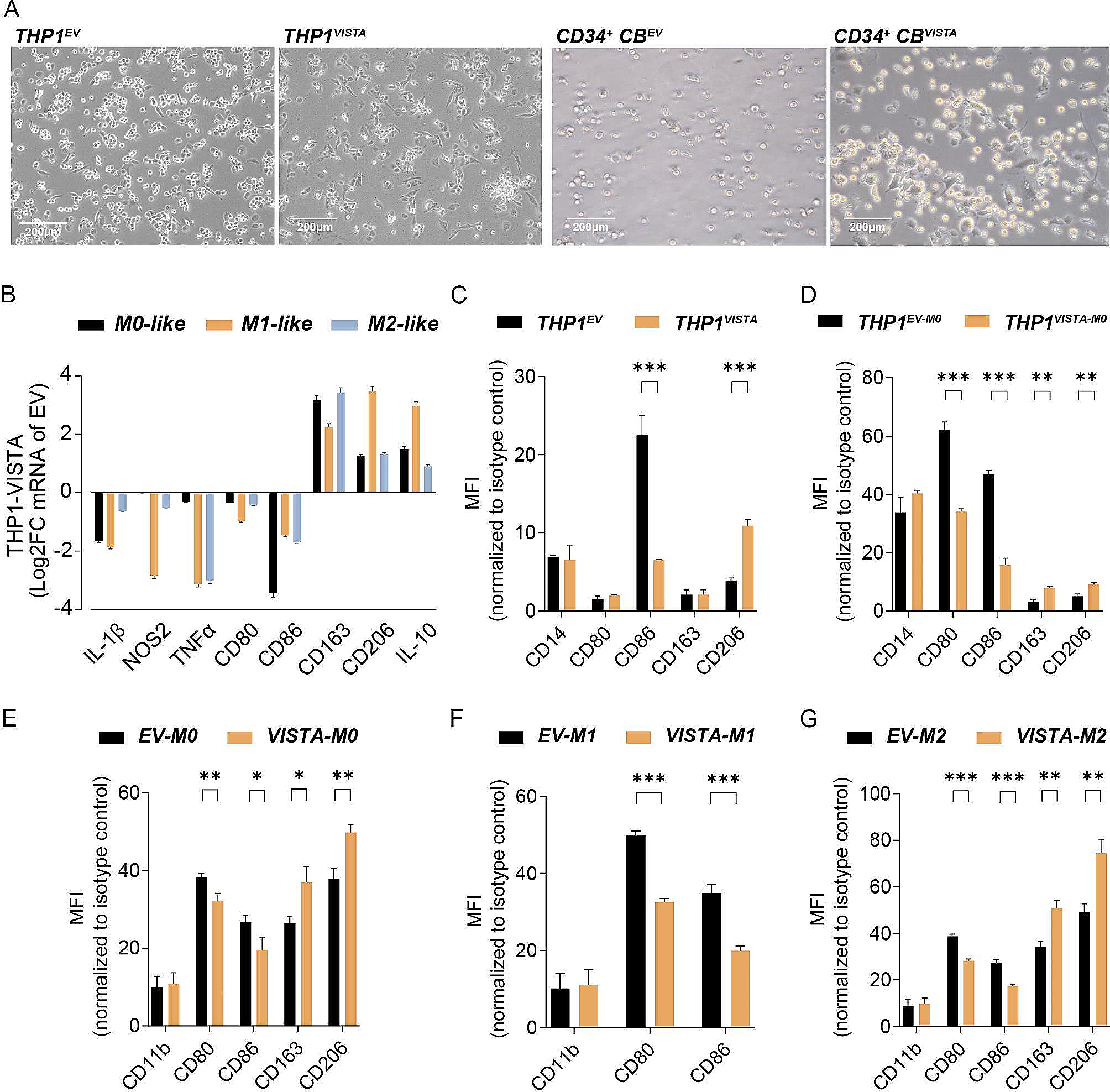
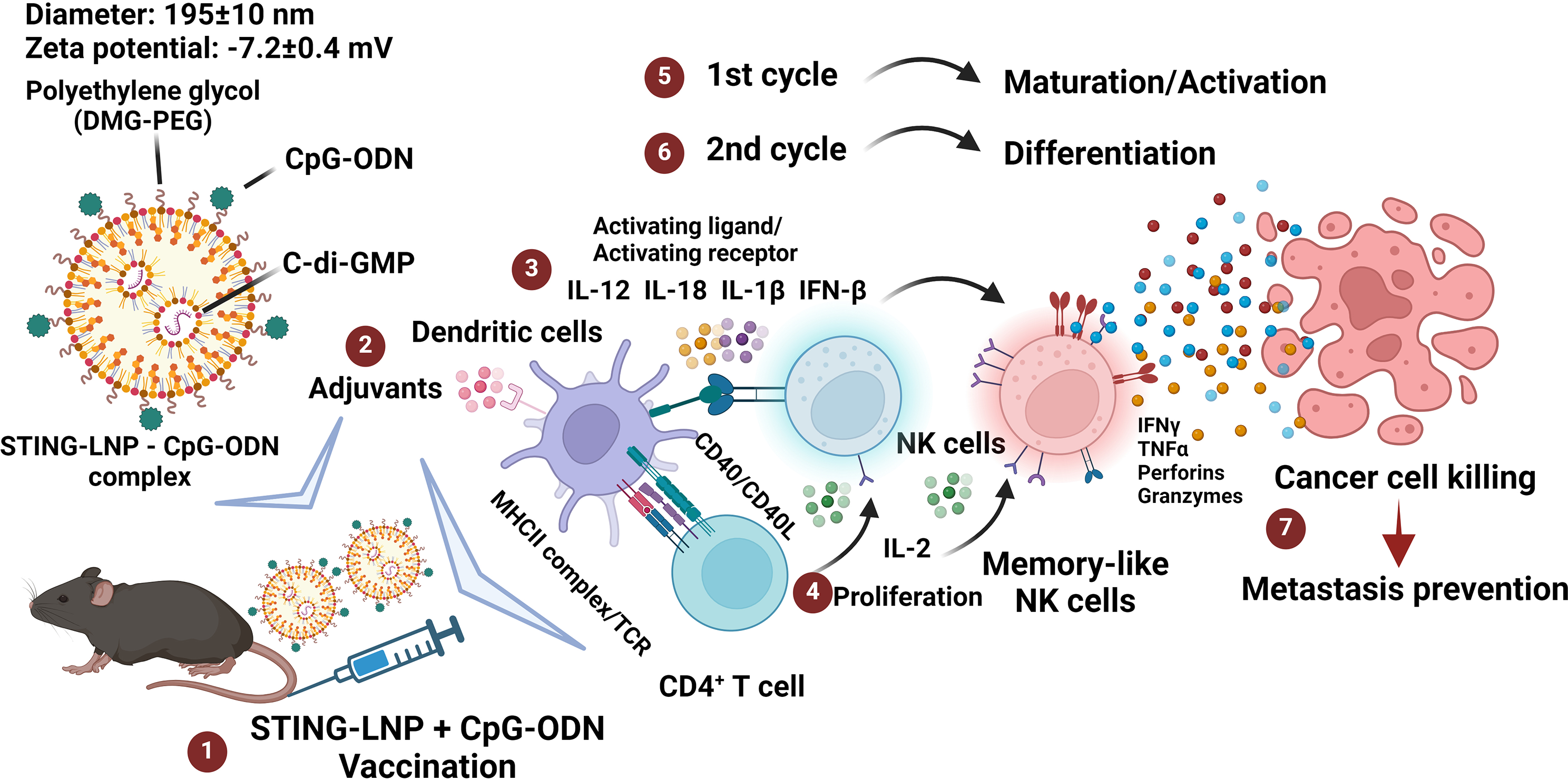
記住我
We performed a survival analysis using the KM plotter online tool [42] to explore the potential association between F-box proteins (Fig. 1A) and overall survival (OS) in lung cancer patients. Our preliminary data indicated that the enhanced expression of certain F-box genes (including FBXL3, FBXL5, FBXL8, and FBXO9) exerted a favorable influence on OS (Supplementary Fig. S1A). Building on these observations, we integrated the tetracycline (Tet)-inducible gene expression system to regulate the specific F-box gene cDNA (as depicted in Supplementary Fig. S1B) in 889DTC cells (KrasG12D /Trp53−/−), which are known for their high metastatic potential [37]. We performed a cell migration assay after confirming the successful expression of these F-box genes by immunoblot analysis (Supplementary Fig. S1C). Overexpression of Fbxo9 significantly reduced cancer cell migration compared with the overexpression of Fbxl3, Fbxl5, or Fbxl8 (Supplementary Fig. S1D and E). However, its overexpression had only a minimal effect on clonogenic survival (Supplementary Fig. S1F and G). These findings prompted further investigation into the biological role of Fbxo9 in various cancer cell lines.
Fig. 1
Suppressive role of FBXO9 in migration, tumor sphere formation and metastasis of lung cancer cells. A Schematic diagram illustrating the role of F-box proteins in CRL1/SCF ubiquitin ligase. B Fbxo9 expression, including that of the Fbxo9-ΔF mutant lacking the F-box domain, is controlled using the Tet-On system. C Co-immunoprecipitation assay identifying the binding capacity between Fbxo9-ΔF and the Skp protein. D Immunoblot analysis of 889DTC cells expressing Fbxo9 and Fbxo9-ΔF, with Gapdh as a loading control. E Representative images and quantitative results of migrated 889DTC cells expressing either Fbxo9 or Fbxo9-ΔF(P < 0.001). F Immunoblot analysis of H1299 cells expressing FBXO9 and FBXO9-ΔF, with GAPDH serving as a loading control. G Representative images (left) and quantitative results (right) of migrated H1299 cells expressing FBXO9 or FBXO9-ΔF (P < 0.0001). H Immunoblot analysis of Fbxo9 knockdown in 889DTC cells using shRNA, with Gapdh as a loading control. I Representative images (left) and quantitative results (right) of migrated 889DTC cells with Fbxo9 knockdown (P < 0.01). J Immunoblot analysis confirming FBXO9 knockdown in H1299 cells using shRNA. K Representative images (left) and quantitative results (right) of cell migration in H1299 cells with FBXO9 depletion (P < 0.0001). L Impact of Fbxo9 knockdown on tumor sphere formation in 889DTC cells (P < 0.0001). M Impact of FBXO9 knockdown on tumor sphere formation in H1299 cells (P < 0.0001). N Ectopic expression of Fbxo9 reduces lung cancer cell metastasis in vivo. Nude mice were injected with 889DTC cells expressing Fbxo9 via the tail vein (n = 6 each group). After two weeks, lung tissues were analyzed. Fluorescence microscopy detected the presence of 889DTC-Tdtomato cells, as indicated by orange fluorescence (left). The number of metastasis nodules was quantified based on fluorescence and statistically analyzed (right) (P < 0.001). O Downregulation of FBXO9 promotes lung cancer cell metastasis in vivo. H1299-Tdtomato cells with stable FBXO9 knockdown were injected into the tail vein of nude mice (n = 6 each group). After 6 weeks, lung tissues were collected for fluorescence microscopy (left) and H&E staining (left). Metastasis nodules were quantified via fluorescence microscopy and statistically analyzed (right) (P < 0.01). All error bars represent the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
We generated a mutated variant of Fbxo9 (known as Fbxo9-ΔF) that lacks the F-box domain (Fig. 1B). The Tet-On gene expression system was used to control the expression of intact Fbxo9 and Fbxo9-ΔF within the cells (Fig. 1B). Our IP assay confirmed that unlike full-length Fbxo9, Fbxo9-ΔF cannot interact with Skp protein (an adaptor subunit of CRL1/SCF ubiquitin ligase) (Fig. 1C). This indicates that Fbxo9-ΔF could not facilitate the assembly of the E3 ligase complex. Subsequently, we introduced full-length Fbxo9 and Fbxo9-ΔF into 889DTC cells and induced their expression using doxycycline (Fig. 1D). Cells expressing full-length Fbxo9 exhibited a significant reduction in migration compared with those expressing Fbxo9-ΔF (Fig. 1E). Similar results were obtained when overexpressing full-length FBXO9 and FBXO9-ΔF in A549 and H1299 lung cancer cells (Fig. 1F and G, Supplementary Fig. S2A-E). Conversely, the depletion of Fbxo9 or FBXO9 using shRNA in 889DTC or H1299 cells significantly increased cancer cell migration (Fig. 1H-K) but had minimal effects on the survival of lung cancer cells (Supplementary Fig. S3A and B). Overall, our observations strongly suggested that FBXO9 serves as a suppressor of lung cancer cell migration but had a limited impact on cancer cell survival.
F-box proteins are implicated in the modulation of cancer stem cell characteristics and play a crucial role in cancer metastasis [28]. The introduction of full-length Fbxo9 significantly reduced the number and size of spheres in 889DTC cells, whereas Fbxo9-ΔF had minimal effect (Fig. 1L). Similar outcomes were observed in the H1299 cell line when FBXO9 and FBXO9-ΔF were overexpressed (Fig. 1M). These findings strongly indicate a significant correlation between FBXO9 and the stem cell-like characteristics of lung cancer cells.
We conducted experiments on mice to study how altering Fbxo9 expression in 889DTC cells affects lung metastasis. We injected the cells into the mice’s tail veins and found that mice with increased Fbxo9 expression had a significantly lower number of metastatic nodules in their lungs compared with that of the control group (Fig. 1N). This indicates that higher Fbxo9 expression can suppress lung metastasis. In another experiment, we introduced H1299 cancer cells with FBXO9 knockdown through tail vein injection. After six weeks, we noticed a noticeable increase in the number of lung metastatic nodules in the FBXO9 knockdown group (Fig. 1O). This result further reinforces the significance of FBXO9 reduction in promoting lung cancer cell metastasis relative to the control group. Collectively, our findings provide evidence that FBXO9 functions as an inhibitor and effectively suppresses lung cancer cell migration, tumor sphere growth, and overall metastasis in cell cultures and mouse models.
FBXO9 interacts with V-ATPaseWe examined the interacting proteins of FBXO9 to elucidate the mechanism underlying the anticancer effects of FBXO9. We established a stable 889DTC cell line expressing a FLAG-tagged Fbxo9-ΔF. Subsequently, we treated the cells with DSP, a chemical crosslinking agent, and performed IP assay and MS to identify the proteins associated with Fbxo9 (Fig. 2A). Remarkably, the absence of the Skp1 binding domain in Fbxo9-ΔF hindered its association with Cullin-1 and the catalytic RBX RING finger subunit, potentially causing substrate entrapment. Consequently, we successfully identified numerous proteins that potentially associate with Fbxo9. The top protein candidates were three members of the heat shock protein family (Hsp90AA1, Hsp90AB1, and HspA8), three subunits of the V-ATPase peripheral V1 domain (Atp6v1a, Atp6v1b2, and Atp6v1f), and all subunits of the TRiC cytosolic chaperonin tailless complex polypeptide one ring complex (Cct1-Cct8) (Fig. 2B). The link between V-ATPase and cancer metastasis progression and drug resistance has been previously established [20, 43]. Therefore, we focused on the constituents of the V-ATPase V1 domain. We hypothesized that that FBXO9 might regulate V-ATPase activity and cancer metastasis through V1 domain ubiquitination. This was tested by transiently transfecting Fbxo9 and separating the V1 domain subunits into HEK293T cells, followed by performing an IP assay to examine the interaction between Fbxo9 and the V1 domain. Immunoblotting revealed that Fbxo9 co-immunoprecipitated with Atp6v1a, while other subunits of the V1 domain exhibited minimal affinity for Fbxo9 (Fig. 2C). Atp6v1a was readily pulled down when Fbxo9 was used as bait (Fig. 2D). This indicated the strong interaction between Fbxo9 and Atp6v1a.
Fig. 2
FBXO9 is closely associated with V-ATPase. A Workflow diagram representing the FBXO9 interactor search process using MS. B List of top proteins identified as potential binders to FBXO9. C Co-IP of different subunits (HA-tagged) of the V-ATPase V1 domain. The V-ATPase V1 domain subunits were immunoprecipitated using anti-HA beads, and the presence of bound FLAG-Fbxo9 was detected using anti-FLAG antibodies. D Co-IP of FLAG-Fbxo9 with HA-Atp6v1a in HEK293T cells. FLAG-Fbxo9 was immunoprecipitated using anti-FLAG antibodies, and the presence of bound HA-Atp6v1a was detected using anti-HA antibodies. E Schematic diagram illustrating the design of full-length HA-tagged ATP6V1A (FL) and a series of truncated, overlapping constructs (left). HEK293T cells were co-transfected with these constructs, followed by IP assay using anti-HA beads. Immunoblots were then probed with both anti-FLAG and anti-HA antibodies to detect the expression of the constructs (right). F Schematic diagram demonstrating the design of full-length FLAG-tagged FBXO9 (FL) and multiple truncated, overlapping constructs. HEK293T cells were co-transfected with these constructs and subsequently immunoprecipitated with anti-FLAG beads. Immunoblotting was conducted using anti-HA and anti-FLAG antibodies to detect the expression of the constructs (right). G Co-IP of the endogenous ATP6V1A with ectopically expressed FLAG-FBXO9 was conducted in HEK293T cells
We subsequently examined the interaction between the human proteins ATP6V1A and FBXO9. We created a series of deletion mutants for ATP6V1A (Fig. 2E) to identify the exact region of ATP6V1A involved in binding to FBXO9. Each of these constructs was individually cotransfected with the FBXO9 construct into HEK293T cells. Subsequent co-IP experiments indicated that amino acid regions 1–83 (1–83 aa) and 230–617 (230–617 aa) play crucial roles in the interaction with FBXO9 (Fig. 2E). A similar approach was employed to map other domains by generating deletion mutants of FBXO9 (Fig. 2F, left). The IP assay emphasized that the C-terminal domain (240–447 aa) of FBXO9 significantly contributed to the interaction with ATP6V1A (Fig. 2F, right). We transfected HEK293T cells with FLAG-FBXO9 and performed IP to detect endogenous ATP6V1A to strengthen our findings. Ectopically expressed FLAG-FBXO9 pulled down a substantial amount of endogenous ATP6V1A compared with the negative control (Fig. 2G). Collectively, these results confirm the interaction between FBXO9 and ATP6V1A, suggesting that FBXO9 may play a role in the function and regulation of V-ATPase.
FBXO9 promotes non-degradative ATP6V1A ubiquitinationWe investigated whether FBXO9 could promote ATP6V1A ubiquitination using a histidine-based ubiquitination assay since FBXO9 is a potential substrate recognition element in CRL1/SCF ubiquitin ligase [34, 35]. As anticipated, the ectopic expression of FBXO9 markedly augmented ATP6V1A ubiquitination. In contrast, the FBXO9-ΔF variant that lacks the essential F-box domain required for complex formation with CRL1/SCF had minimal impact on ATP6V1A ubiquitination (Fig. 3A and B). We reduced the expression of endogenous FBXO9 using shRNA to confirm the involvement of FBXO9 in ATP6V1A ubiquitination. This reduction substantially decreased ATP6V1A ubiquitination (Fig. 3C). We then selectively inhibited the expression of Cullin-1 and SKP1 (scaffold and adaptor proteins of the CRL1/SCF complex, respectively) to gain further insight into the mechanism of FBXO9. The decreased expression of these proteins also resulted in reduced FBXO9-mediated ATP6V1A ubiquitination (Fig. 3D). Taken together, these findings strongly suggest that ATP6V1A is a newly identified substrate of the CRL1/SCFFBXO9 E3 ligase.
Fig. 3
FBXO9 promotes non-degradative ubiquitination of ATP6V1A. A-C Ubiquitination assays in HEK293T cells were performed under different conditions. Co-transfection with HA-Atp6v1a (A) or HA-ATP6V1A (B) and His-Ub, along with Flag-FBXO9 after knockdown of FBXO9 using independent shRNAs (shFBXO9-2# and-3#) (C). Samples recovered with Ni-NTA (top) and whole cell lysate samples (bottom) were subjected to blotting against anti-HA or Flag as indicated. D Involvement of SKP1 and Cullin-1 in FBXO9-mediated ATP6V1A ubiquitination. HEK293T cells were transfected with His-Ub plasmids and shRNA targeting SKP1 or Cullin-1. Cell lysates were subjected to Ni-NTA bead pulldown, and the precipitates were analyzed by immunoblotting. E-G Effect of FBXO9 overexpression on exogenous (E, F) and endogenous (G) ATP6V1A protein levels. Cells were co-transfected with specific plasmids, and immunoblotting was carried out 48 h after transfection. H, I Effect of FBXO9 knockdown on ATP6V1A protein levels. A549 cells (H) and 889DTC cells (I) were depleted of FBXO9 or Fbxo9 using shRNA, and immunoblotting was performed. J, K Impact of FBXO9 expression on ATP6V1A half-life. HEK293T cells were transfected with FLAG-FBXO9 and subsequently treated with CHX for specific time periods, followed by immunoblotting analysis (J). The density of the ATP6V1A band was quantified to determine the ATP6V1A half-life (K). L Effect of neddylation inhibition on the ATP6V1A protein level in HEK293T cells. Cells were treated with MLN4924 for 12 h to inactivate the CRL ubiquitin ligase. Cell lysates were then analyzed by immunoblotting. M–O Ubiquitination of HA-ATP6V1A1 was measured in cells co-expressing FLAG-FBXO9 and different forms of His-Ub, as described in (A-C).
We manipulated the expression of FBXO9 in HEK293T and cancer cells via gene overexpression (Fig. 3E-G) and RNA interference (Fig. 3H and I) to determine the implications of FBXO9-mediated ATP6V1A ubiquitination. The ATP6V1A levels remained largely unaffected regardless of the changes in FBXO9 expression (Fig. 3E-I). Furthermore, ectopic expression of FBXO9 did not increase ATP6V1A degradation in the CHX chase assay, which measures protein turnover (Fig. 3J and K). Additionally, treating cells with MLN4924 (a neddylation inhibitor that deactivates CRL ubiquitin ligases) did not cause significant accumulation of ATP6V1A within cells, although it increased the protein expression of NRF2 (a known substrate of CRL) (Fig. 3L). These findings suggest that the role of FBXO9 in ATP6V1A ubiquitination may not directly affect its expression or protein turnover.
Polyubiquitination typically results from the formation of ubiquitin chains at lysine (K) 48 or K63. K48-linked polyubiquitination mainly signals proteasomal degradation, while K63-linked polyubiquitination often determines protein location, activity, or complex formation [44, 45]. We performed ubiquitination assays using Ub-K48 and Ub-K63 ubiquitin mutants to investigate ATP6V1A polyubiquitination in relation to K48- or K63-linked ubiquitin chains. FBXO9 enhanced K63-linked polyubiquitination of ATP6V1A in HEK293T cells, but not K48-linked polyubiquitination with K48 His-Ub (Fig. 3M). Similarly, co-transfection of HEK293T cells with the Ub-K63R ubiquitin mutant significantly reduced ATP6V1A ubiquitination (Fig. 3N). These findings were also observed in Atp6v1a cells overexpressing Fbxo9 (Fig. 3O). These findings provide evidence that FBXO9 plays a significant role in the K63-linked polyubiquitination of ATP6V1A, suggesting its non-degradative function in cellular processes.
ATP6V1A ubiquitination by FBXO9 suppresses lung cancer cell migration and tumor sphere growth and restricts in vivo metastasisWe used a mutagenesis approach to determine the specific site(s) of FBXO9-mediated ATP6V1A ubiquitination. We mutated ATP6V1A so that all lysine residues were replaced with arginine (ATP6V1A-KR). We also generated a series of overlapping ATP6V1A mutants (Supplementary Fig. S4A). The mutants overlapping with ATP6V1A-KR (ATP6V1A-KR2, ATP6V1A-KR3, and ATP6V1A-KR4) showed partial ubiquitination upon FBXO9 overexpression using a histidine-based ubiquitination assay (Supplementary Fig. S4B). This suggested the presence of potential ubiquitination sites between lysine residue 132 and 414 (Supplementary Fig. S4B). Subsequently, we identified eight specific lysine residues that appeared to be crucial for ATP6V1A ubiquitination. Remarkably, reintroducing lysine residue 393 (K393) into ATP6V1A-KR significantly enhanced ubiquitination signals according to a histidine-based ubiquitination assay (Fig. 4A and Supplementary Fig. S4C). This finding emphasizes the vital role of K393 in FBXO9-mediated ATP6V1A ubiquitination. Interestingly, substituting K393 with arginine in wild-type ATP6V1A to generate the K393R mutant nearly abolished ATP6V1A ubiquitination (Fig. 4B). Furthermore, cross-species analysis demonstrated the conservation of K393 (Fig. 4C). The K393R mutation in Atp6v1a significantly decreased Atp6v1a ubiquitination compared to wild-type Atp6v1a when Fbxo9 was overexpressed (Fig. 4D). This confirmed the ubiquitination site.
Fig. 4
FBXO9-mediated ATP6V1A ubiquitination impairs metastasis of lung cancer cells. A, B Ubiquitination assays were performed in HEK293T cells by co-transfection with full-length HA-ATP6V1A, HA-ATP6V1A-KR (lysine residues removed), HA-ATP6V1A-K393 (K393 reintroduced into the ATP6V1A-KR mutant), or HA-ATP6V1A-K393R (single lysine substitution mutant), in the presence or absence of FLAG-FBXO9. After 48 h, cells were lysed, followed by Ni-NTA bead pull-down and immunoblotting for HA-ATP6V1A. C Alignment of ATP6V1A K393 conservation across species. D Ubiquitination analysis of K393 in HA-Atp6v1a by co-expressing Flag-Fbxo9 and His-Ub with indicated HA-Atp6v1a constructs (WT, K393, or K393R) in HEK293T cells. Cells were lysed after 48 h, followed by Ni-NTA bead pull-down and immunoblotting for HA-Atp6v1a. E V-ATPase complex reconstitution was achieved in H1299 cells by introducing sgRNA-resistant ATP6V1A (wild-type or K393R mutant) using a Tet-Off system. CRISPR was then used to deplete endogenous ATP6V1A, allowing the V-ATPase complex to be reconstituted. Immunoblot analysis confirmed comparable expression levels of the added ATP6V1A and endogenous subunits ATP6V1B and ATP6V1D1, in the experimental group compared to the control cells, after 48 h of doxycycline treatment. F–I Recombinant cell lines H1299-KO-V1A-wt, H1299-KO-V1A-K393R, and H1299-EGFP-sgCtrl were used in a transwell migration assay (F, G) and tumor sphere formation assay (H, I) to evaluate the effects of ATP6V1A ubiquitination on cell migration and tumor sphere growth(P < 0.001). J–N To assess the effects of ATP6V1A ubiquitination on cell migration and tumor sphere growth, V1A-23aa was introduced into A549 cells to inhibit ATP6V1A ubiquitination (J) which was followed by transwell migration assay (K, L) and tumor sphere formation assay (M, N) to evaluate the impact on cell migration and tumor sphere growth, respectively (P < 0.001). O, P Impact of ATP6V1A ubiquitination inhibition by FBXO9 on in vivo lung metastasis was assessed. A549 cells expressing V1A-23 aa were injected into mice via the tail vein. After 6 weeks, the lungs of the mice were harvested for examination and H&E staining. Metastasis nodules were quantified and statistically analyzed using fluorescence microscopy. Error bars represent the mean ± SD. ****P < 0.0001 indicates the significance level
We established H1299 cell lines expressing sgRNA-resistant ATP6V1A in either its wild-type or K393R mutant form using a Tet-Off expression system to further explore the biological implications of the ATP6V1A ubiquitin modification at K393. We selectively depleted endogenous ATP6V1A and reconstituted the V-ATPase complex using CRISPR. Immunoblot analysis confirmed that the ectopic expression levels of ATP6V1A were comparable to those in the control cells and the presence of endogenous subunits such as ATP6V1B and ATP6V1D1 was unaffected by ATP6V1A expression (Fig. 4E). These findings indicate that reconstitution of the V-ATPase complex through ectopic expression of ATP6V1A accurately mimics physiological conditions. Transwell migration and tumor sphere formation assays demonstrated that the ATP6V1A-K393R mutant significantly enhanced cell migration (Fig. 4F and G) and tumor sphere growth (Fig. 4H and I), which is consistent with observations of previous studies showing the augmented effects of FBXO9 depletion (Fig. 1H-K). Furthermore, knockdown of ATP6V1A in cancer cell lines using shRNA inhibited cancer cell migration in mouse and human cell models (Supplementary Fig. S5A-F). These findings suggested a potential connection between FBXO9-mediated ATP6V1A ubiquitination and its inhibitory regulatory effect on the protein.
To gain further insights, we generated cancer cell lines with inducible expression of V1A-23 aa (a peptide that inhibits ATP6V1A ubiquitination) by disrupting the interaction between FBXO9 and ATP6V1A (Supplementary Fig. S6A-D). Cell migration assays and tumor sphere formation experiments revealed that the expression of V1A-23 aa significantly enhanced tumor cell migration (Fig. 4J-L and Supplementary Fig. S7A and B) and tumor sphere formation (Fig. 4M and N, Supplementary Fig. S7C and D) in both the A549 and H1299 cell lines. These data support previous results suggesting that the FBXO9 inhibition enhances the migration phenotype of cancer cells (Fig. 1H-K). We injected H1299 cells expressing V1A-23 aa into the mouse tail vein to assess the effect of FBXO9-mediated inhibition of ATP6V1A ubiquitination on lung metastasis in vivo. Microscopic analysis and hematoxylin-eosin (H&E) staining of mouse lungs at six weeks post-inoculation demonstrated a substantial increase in lung metastatic nodules in the group expressing V1A-23 aa compared with that in the control samples (Fig. 4O and P). This effect was similar to that observed when depleting FBXO9 in the H1299 cell metastasis mouse model. Hence, these in vivo results confirm the potential role of FBXO9 as an inhibitory protein against the migration, tumor sphere growth, and metastasis of lung cancer cells by promoting ATP6V1A ubiquitination.
ATP6V1A ubiquitination mediated by FBXO9 decreases V-ATPase assemblyATP6V1A plays a crucial role as a component of the catalytic head group of the V-ATPase V1 domain and significantly influences lysosomal acidification [9]. We initially reduced FBXO9 expression in A549 lung cancer cells by siRNA transfection to investigate the effect of ATP6V1A ubiquitination on V-ATPase (Fig. 5A). The effect on lysosomal pH was evaluated using LysoTracker Red, a specialized red fluorescent probe that specifically accumulates in acidic organelles such as lysosomes. Notably, we observed a significant increase in red fluorescence intensity within cells upon FBXO9 knockdown (Fig. 5B and C). Similar increases in fluorescence intensity were observed using the FBXO9-knockout H1299 cell line as an additional model to further support these findings. Notably, treatment with BAF (a specific inhibitor of V-ATPase) significantly decreased the red fluorescence intensity in FBXO9-knockout H1299 cells (Fig. 5D and E). This indicated a strong connection between FBXO9 and V-ATPase-regulated lysosomal acidity. Based on this observation, we aimed to alleviate intracellular ATP6V1A ubiquitination in H1299 cells by introducing V1A-23 aa via ectopic expression. This intervention resulted in a notable increase in red fluorescence intensity (Fig. 5F and G), which suggested that FBXO9-driven ATP6V1A ubiquitination impairs V-ATPase complex activity.
Fig. 5
ATP6V1A ubiquitination by FBXO9 impairs V-ATPase assembly. A–C Assessment of lysosomal acidity in A549 cells after FBXO9 knockdown. A549 cells were transfected with FBXO9 siRNA and stained with LysoTracker Red. Plots show the overall LysoTracker intensity per cell (Scale bar = 100 μm, n = 6–8 images from three independent experiments, ****P < 0.0001). D, E Representative images demonstrate LysoTracker Red staining in negative control and FBXO9-knockout H1299 cells. FBXO9-knockout H1299 cells treated with 200 nM bafilomycin A1 (BAF) for 12 h were used as a rescue control. The average staining intensity was quantified as in C. Scale bar = 100 μm, ***P < 0.0001 and ****P < 0.0001. F, G Representative images demonstrate LysoTracker Red staining in H1299 cells ectopically expressing V1A-23 aa. The average staining intensity was quantified using the method described in C. H, I Analysis of V-ATPase assembly in FBXO9-knockdown H1299 cells. FBXO9 was depleted using shRNA, followed by subcellular fractionation. Immunoblot analysis was conducted using antibodies targeting ATP6V1A and ATP6VoD (H). LAMP1 and GAPDH were used as loading controls for membrane and cytosolic proteins, respectively. Ratio of ATP6V1A (n = 3 independent experiments, *P < 0.05, **P < 0. 01) to ATP6VoD in the membrane fraction represents the assembly of the V-ATPase (I). J, K Analysis of V-ATPase assembly in FBXO9-knockout H1299 cells. sgCtrl and FBXO9-knockout H1299 cells were fractionated, followed by immunoblot analysis of ATP6V1A and ATP6VoD (J). Levels of assembled ATP6V1A (K) (n = 3 independent experiments, **P < 0.01) were normalized against ATP6VoD in the membrane fraction. L, M Analysis of V-ATPase assembly in H1299 cells ectopically expressing V1A-23 aa or EGFP is depicted. Representative immunoblots for ATP6V1A, ATP6VoD, LAMP1, and GAPDH are presented (L). The levels of assembled ATP6V1A (M) (n = 3 independent experiments, **P < 0. 01) are measured plotted, and normalized against ATP6VoD in the membrane fraction (M). N, O HEK293T cells were treated with MLN4924 at indicated concentrations for 12 h. Immunoblotting using antibodies against ATP6V1A and ATP6VoD was performed on cytosolic and membrane fractions (N). The levels of assembled ATP6V1A (O) (n = 3 independent experiments, **P < 0. 01) were determined as described in panel (M)
Disassembly and assembly of the V1 and Vo domains of V-ATPase on the vesicular membrane are the primary mechanisms regulating V-ATPase activity [10, 14]. We examined the levels of the assembled V-ATPase V1-Vo holoenzymes on the vesicular membrane to understand how FBXO9-mediated ATP6V1A ubiquitination affects the activity of the V-ATPase complex. This analysis was performed using FBXO9 knockdown or knockout cells. We isolated the cell membrane fraction and quantified the abundance of membrane-associated V1A subunit relative to that of the VoD subunit. This served as an indicator of V1-Vo holoenzyme assembly. Our findings demonstrated a significant increase in the docking level between the V1 and Vo domains in FBXO9-knockdown H1299 cells using shRNA and FBXO9-knockout H1299 cells (Fig. 5H-K). This increase was supported by the presence of ATP6V1A in the membrane components. Additionally, we observed a parallel promotive effect of Fbxo9 knockout on increased vesicular acidification and ATP6V1A localization to the membrane of mouse-derived 889DTC cells (Supplementary Fig. S8A and B). Overall, these findings suggest that the downregulation of FBXO9 expression facilitates V-ATPase assembly, resulting in improved lysosomal acidification.
We blocked ATP6V1A ubiquitination in H1299 cells by introducing V1A-23 aa to investigate the specific role of FBXO9-mediated ATP6V1A ubiquitination in V-ATPase assembly. Interestingly, this approach significantly enhanced the membrane localization of ATP6V1A, which is similar to that observed in FBXO9-depleted cells (Fig. 5L and M). Furthermore, our study demonstrated that neddylation inhibition by MLN4924 promoted the membrane localization of ATP6V1A (Fig. 5N and O). These collective observations provide further evidence that FBXO9-mediated ATP6V1A ubiquitination negatively regulates the activity of the V-ATPase complex by obstructing the assembly of V-ATPase V1-Vo holoenzymes in lysosomal membranes.
HSPA8-mediated cytoplasmic sequestration of ubiquitinated ATP6V1A by FBXO9We further examined how the downregulation of FBXO9 expression aids in the assembly of the V-ATPase holoenzyme on the vesicular membrane. We hypothesized that the underlying mechanisms could be clarified by investigating the proteins that interact with ATP6V1A. Thus, we directed our empirical screening to the cytoplasmic chaperones HSP90AA1, HSP90AB1, and HSPA8, which are associated with ATP6V1A (Fig. 2B). We individually transfected ATP6V1A constructs with HSP90AA1, HSP90AB1, and HSPA8 into HEK293T cells and conducted an IP assay. The results indicated substantial co-IP of HSPA8 with ATP6V1A, whereas the remaining two chaperones showed minimal binding affinity for HSPA8 (Fig. 6A). We then performed domain-mapping experiments and reciprocal IP assays using truncated versions of epitope-tagged HSPA8 and ATP6V1A to investigate the interaction between HSPA8 and ATP6V1A. Immunoblot analysis revealed that the amino acid regions between 1 and 83 aa and 230–617 aa in ATP6V1A were crucial for its interaction with HSPA8 (Fig. 6B). Additionally, a similar strategy confirmed that either the substrate-binding domain (SBD) or the nucleotide-binding domain of HSPA8 was necessary for its interaction with ATP6V1A (Fig. 6C and D). These findings establish a strong correlation between HSPA8 and ATP6V1A expression.
Fig. 6
FBXO9-mediated ATP6V1A ubiquitination leading to cytoplasmic localization facilitated by HSPA8 A Interaction between HSPA8 and ATP6V1A assessed by co-IP assay. HEK293T cells were co-transfected with indicated constructs. After 36 h, co-IP and immunoblot was performed to detect the associated HA-ATP6V1A using anti-HA antibodies. B Mapping the binding domain of ATP6V1A with HSPA8. Different versions of HA-tagged ATP6V1A, including full-length (FL) and truncated constructs, were co-transfected with FLAG-HSPA8 in HEK293T cells. Co-IP was performed to assess the interaction, and the presence of bound FLAG-HSPA8 was detected using anti-FLAG antibodies. C, D Mapping the binding domain of HSPA8 with ATP6V1A. Different forms of FLAG-tagged HSPA8 constructs, including full-length (FL) and truncated versions, were designed and shown (C). HEK293T cells were co-transfected with HA-ATP6V1A and the corresponding FLAG-tagged HSPA8 constructs. Co-IP assay was performed using anti-FLAG beads to assess the interaction between HSPA8 and ATP6V1A. The presence of bound HA-ATP6V1A was detected using anti-HA antibodies (D). E Impact of FBXO9 knockdown on the interaction between ATP6V1A and HSPA8. HEK293T cells pre-treated with FBXO9-shRNA were co-transfected with HA-ATP6V1A and FLAG-HSPA8. After 36 h, a co-IP assay was performed to evaluate the effect of FBXO9 depletion on the interaction between ATP6V1A and HSPA8. F Effect of FBXO9 knockout on the interaction between endogenous ATP6V1A and HSPA8. FBXO9-knockout H1299 was used and a co-IP assay was performed using anti-ATP6V1A antibody. The presence of the bound endogenous HSPA8 was detected using anti-HSPA8 antibodies. G Role of FBXO9-mediated ubiquitination in modulating the interaction between ATP6V1A and HSPA8. HEK293T cells were co-transfected with FLAG-HSPA8 and either HA-ATP6V1A or HA-ATP6V1A-K393R. The co-IP assay was then conducted to evaluate the interaction between HSPA8 and ATP6V1A. H, I Impact of HSPA8 depletion on lysosomal acidity in A549 cells. HSPA8 was knocked down using shRNA (H, upper). The cells were stained with LysoTracker Red and fluorescence was measured (H, lower). Staining intensity was quantified as outlined in Fig. 5C (I). The scale bar is 50 µM; (P < 0.0001). J, K Impact of HSPA8 depletion on V-ATPase assembly in A549 cells. HSPA8-knockdown cells were fractionated, and immunoblotting was performed using antibodies against ATP6V1A and ATP6VoD (J). Membrane and cytosolic proteins were assessed with LAMP1 and GAPDH as loading controls, respectively. Levels of assembled ATP6V1A were measured and normalized to ATP6VoD in the membrane fraction (K). Results are from three independent experiments (P < 0.0001). L, M Blocking the interaction between HSPA8 and ATP6V1A enhances lysosomal acidity in A549 cells. Cells expressing the A8-40 aa peptide were stained with LysoTracker Red and fluorescence was observed (L). The average staining intensity was quantified as outlined in Fig. 5C. Scale bar = 100 µM. ****P < 0.0001. N, O Blocking the interaction between HSPA8 and ATP6V1A promotes V-ATPase assembly. A549 cells expressing the A8-40 aa peptide were fractionated. Immunoblot analysis was then performed to assess the level of V-ATPase in the cells (P < 0.05), as described in Fig. 6J and K
shRNA was used to downregulate FBXO9 expression in HEK293T cells to explore the effect of FBXO9 on the interaction between HSPA8 and ATP6V1A. Subsequently, we co-transfected HSPA8 and ATP6V1A and then performed IP assay to assess their interactions. Immunoblot analysis revealed a decrease in the binding affinity of HSPA8 for ATP6V1A upon FBXO9 depletion (Fig. 6E). Furthermore, we used an ATP6V1A-specific antibody to immunoprecipitate ATP6V1A and detect co-immunoprecipitated endogenous HSPA8 in FBXO9-knockout H1299 cells. FBXO9 depletion significantly inhibited the interaction between ATP6V1A and HSPA8 (Fig. 6F), which indicates that FBXO9 has a substantial role in facilitating this interaction. We then transfected HSPA8 with wild-type ATP6V1A or ATP6V1A-K393R in HEK293T cells to further support this hypothesis. The IP assay confirmed that the ATP6V1A-K393R mutant exhibited a diminished binding capacity towards HSPA8 compared with that of the wild-type form (Fig. 6G). Collectively, these results established that FBXO9-mediated ubiquitination of ATP6V1A enhances its interaction with HSPA8 in the cellular environment.
We investigated the functional implications of the interaction between ATP6V1A and HSPA8 by inhibiting HSPA8 expression in A549 cells using shRNA and examining the effect of HSPA8 depletion on lysosomal pH changes using LysoTracker Red. A significant increase in red fluorescence intensity was observed within the cells upon reduction in HSPA8 levels (Fig. 6H and I). This observation strongly indicated that HSPA8 plays a crucial role in regulating lysosomal acidification, which is primarily controlled by V-ATPase. We performed shRNA-mediated knockdown of HSPA8 in A549 cells to evaluate V-ATPase assembly at the vesicular membrane to understand the mechanism underlying this regulation. Remarkably, A significant increase in the relative abundance of the V1 domain docked with the Vo domain was observed when HSPA8 was reduced compared to that in the control cells (Fig. 6J and K). These findings strongly support the idea that HSPA8 negatively regulates V-ATPase assembly within cells. Based on the observed interaction between HSPA8 and ATP6V1A, we hypothesized that HSPA8 sequesters ATP6V1A in the cytosol to hinder its localization to the vesicular membrane. This was investigated by designing constructs producing peptides of varying lengths (HSPA8-25 aa, 40 aa, 50 aa, and 60 aa) that target the SBD of the HSPA8 protein (Supplementary Fig. S9A). IP assays confirmed that most of these HSPA8 peptides (except for HSPA8-60 aa) effectively disrupted the ATP6V1A and HSPA8 interactions in the cellular environment (Supplementary Fig. S9B). Subsequently, HSPA8-40 aa was selected, and an A549 cell line with ectopic expression of this peptide was established. LysoTracker Red staining and subcellular fractionation revealed a significant increase in red fluorescence intensity (Fig. 6L and M) and ATP6V1A protein localization to the vesicular membrane (Fig. 6N and O). These findings strongly support the hypothesis that FBXO9-mediated ATP6V1A ubiquitination facilitates its interaction with the chaperone protein HSPA8, thus leading to cytosolic sequestration and V-ATPase assembly inhibition.
FBXO9 inhibits wnt signaling and epithelial-mesenchymal transition by ubiquitinating ATP6V1AV-ATPase is a crucial enzyme that plays a significant role in initiating cancer signaling pathways (including Wnt and Notch) by regulating vesicular acidification [18, 19]. The Wnt/β-catenin pathway is particularly important for cancer development [46]. Our investigation aimed to understand the impact of FBXO9 on the Wnt/β-catenin pathway in lung cancer cells. Silencing FBXO9 in A549 cancer cells by siRNA transfection substantially increased the expression of active β-catenin (the primary effector of the canonical Wnt signaling pathway), C-Myc and cyclin D1 (both important downstream targets of this pathway) upon FBXO9 depletion (Fig. 7A). Fbxo9-knockout 889DTC cells showed elevated levels of active β-catenin, C-Myc, and cyclin D1 (Fig. 7B). This was consistent with the results observed in FBXO9-knockdown A549 cells. Remarkably, reintroducing the entire length of Fbxo9 into these Fbxo9-deficient cells restored decreased levels of active β-catenin, C-Myc, and cyclinD1. However, reintroduction of the Fbxo9-ΔF mutant failed to produce the same effect (Fig. 7B). These findings suggest a negative correlation between FBXO9 expression and Wnt/β-catenin signaling activation, which is dependent on the ligase activity of FBXO9. As previous experiments showed that FBXO9 suppresses V-ATPase through ATP6V1A ubiquitination, we sought to explore the relationship between FBXO9 and V-ATPase in the regulation of Wnt/β-catenin signaling in lung cancer cells (Fig. 5). Therefore, Fbxo9-knockout 889DTC cells were treated with BAF. The inhibition of V-ATPase by the spec
留言 (0)