
記住我




We generated a total of 26 WGMS datasets from milk somatic cells from sixteen cows with S. aureus subclinical mastitis and ten healthy control cows (Supplementary Table S1 in Additional file 1). The vast amounts of data (~ 294 million uniquely mapped reads) obtained from each sample revealed ~ 12,215 million cytosines with an average of 27× coverage (ranging from 21.8× to 32.8×) per sample which were used to identify genome-wide DNA methylation cytosine sites (Supplementary Table S2 in Additional file 1). Globally, the average methylation level was 75.28, 0.21, and 0.18% for cytosine in the context of CpG, CHG, and CHH, respectively (where H represents A or T or C). The global DNA methylation level of CpG sites is consistent with data on other somatic tissues in cattle [39], in human [40], and in mouse [41]. Moreover, the global DNA methylation level of CHG sites is consistent with non-CpG methylation recorded in non-brain tissues of cattle (ranged from 0.2 to 0.8%) [39, 42].
We also characterized the DNA methylation patterns in specific functional genomic regions to understand their dynamics during S. aureus subclinical mastitis. The DNA methylation of CpG sites showed a concave change around CpG island (CGIs) with a downward trend in CGI shores and low level at CGIs (28.74% on average) (Fig. 1A). On the contrary, the CHG and CHH sites showed a slight increase in their methylation levels at CGIs (0.29 and 0.21%, respectively) compared with adjacent regions (Fig. 1A). The methylation level plot among gene features revealed the classic downward trend of CpG sites with a gradual decrease in methylation at the promoter to reach the lowest level in the first exon (Fig. 1B). In addition, CHG and CHH sites showed relatively stable and extremely low methylation level among gene regions, but was slightly higher at exons (Fig. 1B).
Fig. 1
DNA methylation landscape at important genomic functional regions indicating their possible association with gene expression. A The distribution of DNA methylation at regions in relation to CpG island (CGI). L: left, R: right. B The DNA methylation level at genetic regions illustrating the classic valley-like change of DNA methylation level (CpG sites) around transcription start sites (TSS). C–E The scatterplot and fitting curves of DNA methylation level of regulatory regions, including promoters (C), first exons (D), and first introns (E), and expression level of genes. The higher the expression level of genes, the lower methylation level of their regulatory regions. F The methylation level trends around genes grouped by ranked gene expression level. Up‑region and down-region represent the upstream and downstream region of genes, respectively. G An enlarged view of figure F near TSS, showing the steeper methylation level trend around TSS for genes with higher expression levels
We then calculated the average methylation levels of promoters and other gene features for each gene (average value of all qualified CpG sites in corresponding regions) followed by integration analysis of the methylome and transcriptome data with MethGET [43]. The CpG methylation level of promoters showed significant but weak inverse correlation with gene expression level at the genome-wide scale (Pearson’s R = − 0.194, P = 3.76 × 10−165) (Fig. 1C, Supplementary Fig. S1A in Additional file 2), which is consistent with previously reported effects of promoter methylation on transcriptional repression [20, 44]. The general CpG methylation levels of both first exons and first introns also showed significant but weak inverse correlation with gene expression levels at the scale of the whole-genome (Pearson’s R: − 0.291 and − 0.173, P = 0 and 1.60 × 10−122, respectively) (Fig. 1D, E, Supplementary Fig. S1B-C in Additional file 2). This inverse correlation relationship has also been found in human and other model animals which links the methylation at first exon and first intron to possible effects on transcriptional silencing [45, 46]. The valley-like changes of CpG sites at regulatory regions, including promoter, first exon and first intron, were steeper as gene expression levels rise (Fig. 1F), especially around the 2000 bp up- and downstream of transcriptional start sites (TSS) (Fig. 1G). Besides, no significant correlation was found between the gene expression levels and the CpG methylation levels of other gene regions (inner exons, inner introns, and whole gene body) (Supplementary Fig. S1D-F in Additional file 2).
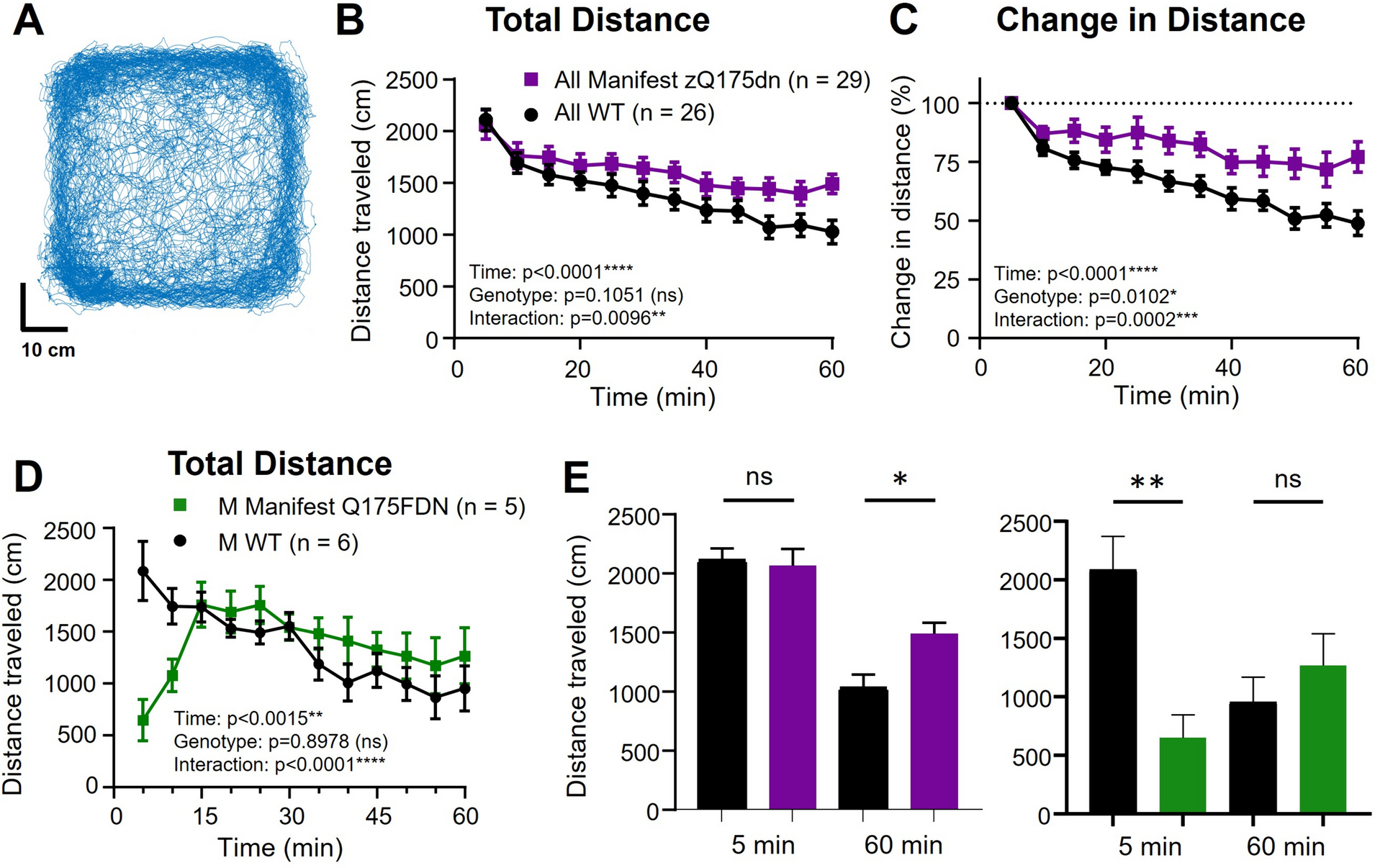
DNA methylation alterations during S. aureus subclinical mastitisWe compared the DNA methylation of cytosine sites between S. aureus-positive (SAP) and healthy control (HC) groups at different layers to investigate the DNA methylation alterations during S. aureus subclinical mastitis. Firstly, the global methylation level of CpG sites of SAP group was significantly higher than HC group at a scope of each chromosome (Chr) (Fig. 2A shows Chr11 only, Supplementary Table S3A in Additional file 1) as well as whole genome (Fig. 2B, Supplementary Table S3B in Additional file 1) (FDR < 0.05). This significant higher methylation level of CpG sites in SAP group was also seen at genomic functional regions, including gene features, CGIs, CGI shores, and CGI shelves (Supplementary Table S3C in Additional file 1, Supplementary Figs. S2-S3 in Additional file 2). However, the difference in methylation levels of CHG and CHH sites between SAP and HC group was not significant at genome-wide scale (Supplementary Fig. S4 in Additional file 2), as well as in most genomic functional regions (FDR > 0.05) (Supplementary Table S3C in Additional file 1) except CHG sites which showed significantly higher expression in SAP group at CGI (FDR = 0.025) (Supplementary Fig. S5 in Additional file 2).
Fig. 2
Comparison of DNA methylation status between S. aureus-positive (SAP) and healthy control (HC) groups revealed abundant alterations. A A DNA methylation landscape of chromosome 11 (Chr11) illustrating the general methylation status and various DNA methylation alterations. A 50-kb-long window was used to count the corresponding information per track. B Comparison of global methylation level of CpG sites between SAP and HC groups revealed significantly higher global methylation level of SAP group. ***: significant difference (P value = 0.00076). C The distribution of DMCs per chromosome (Chr). The number on the top of each bar represents the percentage of hyper-methylated DMCs to total DMCs located in the corresponding Chr. D The distribution of DMCs in the context of CpG, CHG, and CHH among genomic functional regions. E The count of DMCs collocated in repeat elements, including short and long interspersed retrotransposable elements (SINE and LINE), long terminal repeat retrotransposons (LTR), and DNA transposons (DNA). MHBs: methylation haplotype blocks; dMHBs: differential MHBs; DMCs: differentially methylated cytosines which were counted in the context of CpG, CHG, and CHH, respectively; Hyper/hypo: DMCs were hyper-/hypo-methylated in SAP group compared to HC group; CGI: CpG island. Detailed data on the depicted findings are found in Supplementary Tables 3–6
Furthermore, we compared the methylation of each cytosine between SAP and HC group using MethylKit [47], and identified a total of 3,328,843, 7255, and 20,358 differentially methylated cytosine sites (DMCs) in the context of CpG, CHG, and CHH, respectively (q-value < 0.05 and |methylation difference|> 20%) (Supplementary Tables S4-S5 in Additional file 1). Consistent with the globally higher methylation of SAP group, 80.73% of CpG-DMCs and 61.6% of CHG- and CHH-DMCs were hyper-methylated in SAP group compared with HC group (q-value < 0.05, methylation difference ≥ 20%) (Fig. 2C). At Chr scale, Chr11 harbored the most DMCs (n = 162,144, 4.83%), followed by Chr19 (n = 157,932, 4.70%) and Chr5 (n = 155,308, 4.62%), while the least DMCs (n = 56,880, 1.69%) were found on Chr28 (Fig. 2C, Supplementary Table S6A in Additional file 1). The annotation of DMCs to gene features revealed that 69% DMCs were located in intergenic regions (Supplementary Table S6B in Additional file 1). Also, 1,148,481 CpG-, 2295 CHG-, and 5987 CHH-DMCs were located in genes and their 2 kb upstream (promoters) and 2 kb downstream regions. Out of these, more DMCs were located in introns (n = 887,913, 76.76%) followed by exons (n = 112,914, 9.76%) (Fig. 2D). Besides, we found 72,855 (6.30%), 11,599 (1.00%), and 308,719 (26.69%) DMCs in promoters, first exons, and first introns, respectively. 3′UTR regions harbored more DMCs than 5′UTR. However, 5′UTR had the highest density of DMCs (6.15 DMCs/100 bp), followed by first exon (2.52 DMCs/100 bp) and 3′UTR (1.90 DMCs/100 bp) (Supplementary Table S7 in Additional file 1).
On the other hand, we identified a total of 13,358 genes harboring at least 1 DMC in any of three contexts, and 76.58% of them (n = 10,230) harbored greater than 20 DMCs (Supplementary Table S7 in Additional file 1). Integration with transcriptome data revealed that 3599 out of 13,358 genes harboring DMCs were identified as differentially expressed genes (DEGs) between SAP and HC group (|log2FC|> 1 and FDR < 0.05). The DEGs harboring DMCs (n = 3599) were significantly enriched in 20 gene ontology (GO) terms and 35 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways mostly having immune- or disease-related functions (Supplementary Table S8A in Additional file 1). Interestingly, DEGs harboring DMCs at their first exon (n = 431) were significantly enriched in immune-related KEGG pathways, including cytokine-cytokine receptor interaction (bta04060), Staphylococcus aureus infection (bta05150), and viral protein interaction with cytokine and cytokine receptor (bta04061) (Supplementary Table S8B in Additional file 1). In addition to immune- or disease-related functional annotations, DEGs harboring DMCs in the promoter region (n = 2735) were also enriched in KEGG pathways related to metabolism processes, such as fatty acid metabolism (bta01212), propanoate metabolism (bta00640), and fatty acid degradation (bta00071) (Supplementary Table S8C in Additional file 1). This observation suggests that the methylation alterations may be involved in the regulation of mammary gland responses during S. aureus subclinical mastitis, and the DNA methylation at different genetic regions may play different roles.
DNA methylation alterations of repeat elements in response to S. aureus subclinical mastitisAmong all identified DMCs, we found that 28.96% CpG-DMCs (964,144), 41.60% CHG-DMCs (3018), and 43.63% CHH-DMCs (8883) were located in repeat elements (REs) (Fig. 2D). The DMCs were mainly located in non-long terminal repeat retrotransposons, including short interspersed nuclear elements (SINEs) and long interspersed nuclear elements (LINEs), followed by long terminal repeat (LTR) retrotransposons and DNA transposons (Fig. 2E). To further explore the DNA methylation status of REs and their possible changes during S. aureus subclinical mastitis, we used a random forest-based algorithm to calculate the methylation levels of LINE (LINE-1 elements) and SINE (tRNA-derived SINEs), which are the most abundant retrotransposons in the bovine genome. After filtering, a total of 321 LINE-1 elements and 217 tRNA-derived SINEs harboring at least two CpG sites were retained (Supplementary Table S9A-B in Additional file 1). In general, tRNA-derived SINEs had higher CpG density (2.22 CpG/100bp) than LINE-1 elements (0.94 CpG/100bp) (Supplementary Fig. S6 in Additional file 2). However, LINE-1 elements had generally a higher methylation level than tRNA-derived SINEs; their median methylation levels were 60 and 55%, respectively. As shown in Fig. 3A, LINE-1 elements in SAP group showed clearly higher density and methylation level (> 60%) compared to HC. In addition, 96 LINE-1 elements and 23 tRNA-derived SINEs showed significant difference in their methylation levels between SAP and HC group (FDR < 0.05), which also correlated significantly with S. aureus subclinical mastitis status (FDR < 0.05 and |cpb|> 0.3) (Supplementary Table S9c in Additional file 1). Among them, twelve LINE-1 elements and nine tRNA-derived SINEs were collocated with protein-coding genes, and the collocated genes of one LINE-1 (L1_Art, Chr2:135290744-135291147) and three tRNA-derived SINEs (CHR-2A (Chr1:64176302-64176611), CHRL (Chr28:25046248-25046370), and CHR-2B (Chr25:21749299-21749545)) were significantly differentially expressed between SAP and HC group. The methylation status of L1_Art, CHR-2A, and CHRL showed significantly strong and positive correlation with the expression levels of their overlapped genes (PADI4, ARHGAP31, and CCAR1, respectively) (Fig. 3B, C). It is worth noting that L1_Art and CHRL harbored the TSS of PADI4 and CCAR1, respectively. This suggests that the methylation changes of these REs, such as L1_Art, CHR-2A, and CHRL, may be a possible mechanism underlying the gene expression changes of their overlapped genes during S. aureus subclinical mastitis.
Fig. 3
Comparison of the methylation levels of repeat elements between S. aureus-positive (SAP) and healthy control (HC) groups. A The methylation level distribution of LINE-1 (L1) and tRNA-derived SINE (SINE/tRNA) in SAP and HC groups. B, C The methylation level of CHRL (a tRNA-derived SINE, Chr28:25046248-25046370) (B) and L1-Art (a LINE-1, Chr2:135290744-135291147) (C) and the expression level of their overlapped genes, CCAR1 and PADI4, per sample, revealing significant differences between SAP and HC groups as well as positive correlation between methylation level and expression level of their overlapped genes. Detailed data on the depicted findings are found in Supplementary Table 9
Methylation haplotype blocks responding to S. aureus subclinical mastitisTo better identify small regions with abundant DNA methylation alterations, we used MONOD2 to identify MHBs that captures the co-methylation status of adjacent CpG sites exhibiting highly coordinated methylation [48]. Using a linkage disequilibrium (r2) cutoff of 0.5 and minimum 3 CpG sites per block, we identified a total of 750,583 MHBs. The majority of CpG sites within the same MHB were tightly coupled (r2 ~ 1). The MHBs had a median length of 33.0 bp (range from 6 to 388 bp) and a median CpG density of 10.34 CpG/100 bp, which represents about 0.91% of the bovine genome (Supplementary Fig. S7A-B in Additional file 2). About three quarters of MHBs (576,014) were located in the intergenic region, while about 40% (294,672) were overlapped with transcripts. MHBs were also widely distributed in known regulatory regions, such as promoters, CGIs, exons, and introns (Supplementary Fig. S7C in Additional file 2). The overlapping of MHBs with different genomic functional regions suggests their potential to represent a distinct type of genomic feature.
To allow the direct comparison of the methylation status of MHBs between groups, we calculated the methylation haplotype load (MHL) for each MHB which was defined as the normalized fraction of methylation haplotypes at different lengths [48]. We found a total of 153,783 MHBs with significant differences between SAP and HC groups (|MHL difference|> 20% and FDR < 0.05), and are here referred to as differential MHBs (dMHBs) (Supplementary Table S10A in Additional file 1). Among them, 120,033 dMHBs (78.05%) showed higher methylation levels in SAP group. The dMHBs were able to capture clearly the initial classification of SAP and HC groups, as exampled by the most variable dMHBs in Fig. 4A. The length of dMHBs, ranged from 6 to 318 bp, with median length of 53 bp and median CpG density of 6.90 CpG/100 bp (Fig. 4B). The dMHBs harbored a total of 624,957 CpG-DMCs, accounting for 18.77% of total CpG-DMCs. The dMHBs showed similar distribution characteristics with DMCs. The majority of dMHBs (100,141, 65.12%) were found in intergenic regions while 53,876 dMHBs were overlapped with genes (Fig. 4C). The dMHBs also showed significant enrichment in functional regulatory and genic regions, such as exons, introns, first intron, downstream region, first exon, promoter, 3′UTR, and 5′UTR (P value < 0.001) (Fig. 4D). In addition, 30,938 and 50,223 dMHBs were collocated with QTLs for immune capacity and mastitis, respectively (Fig. 4C). It is worth noting that some QTLs coved a very large region and harbored many dMHBs, such as a QTL (#10446, Supplementary Table S10B in Additional file 1) for somatic cell score, which is about 61.3 Mbp long and harbors 8436 dMHBs. Since the extremely long QTLs are usually identified by linkage methods using low-density microsatellites marker maps, we further checked the QTLs with less than one Mbp length and found 73 immune capacity QTLs and 181 mastitis QTLs harboring 355 and 1334 dMHBs, respectively (Supplementary Table S10B in Additional file 1). Among them, 13 dMHBs harbored SNP markers, including three, two, seven, and one dMHBs harboring SNPs for clinical mastitis, SCC, somatic cell score, and eosinophil number, respectively. For instance, Chr1:110158892:110158992 which collocated with exon 3 of PTX3 harbored two SNPs for somatic cell score (QTL #219865 and #219866). Besides, most of the CpG sites in the same dMHB were tightly linked DMCs (r2 ~ 1) which also showed clear differences in their methylation levels between SAP and HC groups (Fig. 4E).
Fig. 4
The dMHBs and the potential effects of DNA methylation at regulatory regions on gene expression. A Heap map of top 50 most variable dMHBs. B The length and CpG density distribution of dMHBs. C Co-localization of dMHBs with genomic functional regions. CGI: CpG islands. D Enrichment of dMHBs at genomic functional regions. E An example of dMHB (Chr29:47439984:47440080) showing the coordinated methylation of CpG sites in the same dMHB. F Global relationship (significant negative correlation) between promoter methylation level and gene expression level. Each dot symbolizes a specific gene. Blue dots indicate that gene expression and methylation level changes for the corresponding gene were not statistically significant (Gaussian Mixture Model p > 0.005). Conversely, red dots represent differential genes with significant changes in both gene expression and methylation levels of their promoters (Gaussian Mixture Model p < 0.005). Red dots out of gray shadow represent differential genes with significant changes in the methylation level of promoter (greater than 10% changes) and gene expression level (|log2FC|≥ 1) between S. aureus-positive (SAP) and healthy control (HC) groups. G The top 10 most significantly enriched known motifs for transcription factors in hyper- and hypo-methylated dMHBs located at regulatory regions and significantly associated with gene expression (GE-dMHBs). H, I Examples of de novo (discovered) motifs in hypo-methylated GE-dMHBs (hypo_VGGAAR) (H) and hyper-methylated GE-dMHBs (hyper_CNGGRA) (I), showing high similarity with known motifs for transcription factors. Detailed data on the depicted findings are found in Supplementary Tables 10, 11 and 14
DNA methylation at regulatory regions as potential regulators of gene expressionWe found significant but weak inverse correlation between the changes in global CpG methylation levels of regulatory regions, including promoter, first exon, and first intron, and corresponding gene expression level changes between SAP and HC groups at genome-wide scale (P < 5 × 10−8) (Fig. 4F, Supplementary Fig. S8 in Additional file 2). Additionally, we identified 882, 1188, and 870 genes with significant changes in their gene expression levels and CpG methylation levels of promoter, first exon, and first intron, respectively, and here referred to as differential genes (|log2FC|> 1, |CpG methylation difference|> 10% and P value < 0.005) (Supplementary Table S11 in Additional file 1). The differential genes were significantly enriched in GO terms and KEGG pathways related to key functions of immune responses and diseases, such as Staphylococcus aureus infection (bta05150), antigen processing and presentation (bta04612), cytokine-cytokine receptor interaction (bta04060), positive regulation of immune system process (GO:0002684) among others, suggesting the potential involvement of DNA methylation at these regulatory regions in the host response to defense against S. aureus infection (Supplementary Table S12 in Additional file 1). Interestingly, the CpG methylation level of the regulatory regions of more than 50% of the differential genes (promoter: 58.28%, first exon: 50.84%, first intron: 66.09%) were significantly correlated their gene expression levels (|Spearman’s rho|> 0.3 and FDR < 0.05, Supplementary Table S11 in Additional file 1). In particular, majority of them had hyper-methylated promoters (n = 286), first exons (n = 300), or first introns (n = 347) which were significantly negatively correlated with the downregulated expression of their corresponding genes. This further revealed that DNA methylation in regulatory regions may mediate gene repression to regulate the immune responses of the mammary gland during S. aureus subclinical mastitis.
Therefore, we selected the dMHBs that overlapped with promoters (n = 1261, pro-dMHBs), first exons and 5′UTR (n = 526, FE-dMHBs), and first introns (n = 6921, FI-dMHBs) of DEGs to further investigate their possible regulatory roles in gene expression during S. aureus subclinical mastitis. We used Spearman’s rank correlation coefficient (rho) to correlate each dMHB to its corresponding DEG. We found that the methylation status of 6435 dMHBs (933 pro-, 400 FE-, and 5250 FI-dMHBs) were significantly correlated with the expression levels of their corresponding DEGs (|rho|> 0.3 and FDR < 0.05), here referred to as gene expression correlated dMHBs (GE-dMHBs) (Supplementary Table S13 in Additional file 1). Among them, the majority of GE-dMHBs, including 76.85% of pro-GE-dMHBs (n = 717), 85.50% of FE-GE-dMHBs (n = 342), and 71.37% of FI-GE-dMHBs (n = 3747) had significant negative correlations between their methylation status and the expression levels of their corresponding DEGs (rho < − 0.3 and FDR < 0.05). In particular, the negatively correlated hyper-methylated GE-dMHBs and their corresponding downregulated DEGs accounted for the largest proportion (i.e., 533 pro-, 137 FE-, and 627 FI-GE-dMHBs).
To explore the possible effects of GE-dMHBs on transcriptional factor (TF) binding regions, we performed a motif search of expressed TFs in 6435 GE-dMHBs, including 4999 hyper- and 1436 hypo-methylated GE-dMHBs. Firstly, 144 and 67 known motifs were significantly enriched in hyper- and hypo-methylated GE-dMHBs, respectively (FDR < 0.05) (Fig. 4G, Supplementary Table S14A in Additional file 1). The most significantly enriched motifs, such as the Fork head/winged helix factors (FOXK1, FOXK2, FOXL1, FOXP1, FOXA3, and others), the TEA domain factors (TEAD1-TEAD4), and the tryptophan cluster factors (EHF, ETV1, ETV4, ELF1, ELF3, and others), have been implicated in the regulation of the expression of a wide range of genes [49, 50]. The enriched SP4, EGR1, and KLF4 TF motifs have been found to regulate gene expression through binding to CpG-rich promoters [51, 52]. Additionally, the discovery of de novo motifs by running Dreme from MEMEs [53] revealed 23 and 13 novel motifs within hyper- and hypo-methylated GE-dMHBs, respectively (Supplementary S14B-C, Supplementary Fig. S9 in Additional file 2). For instance, hypo_VGGAAR and hyper_CNGGRA, the most significantly enriched motifs discovered in hypo- and hyper-methylated GE-dMHBs, respectively, showed high similarity with known STAT domain factors (STAT1 and STAT3, respectively) (Fig. 4H, I). Therefore, the enrichment of known and de novo TF motifs in GE-dMHBs suggests the potential effects of GE-dMHBs on TF binding events and thereby transcriptional activities.
Potential functions of GE-dMHBs in response to S. aureus subclinical mastitisWe applied functional enrichment analysis to GE-dMHBs (their overlapped and significantly correlated DEGs) to explore their potential roles during S. aureus subclinical mastitis using clusterProfiler [54]. GE-dMHBs that negatively correlated with their overlapped DEGs were enriched in more functional GO terms and KEGG pathways than those with positive correlations. Among them, 3604 hyper-methylated GE-dMHBs (they were negatively correlated with their overlapped 816 downregulated DEGs) were significantly enriched in 38 GO terms (28 biological process (BP) (Fig. 5A), one molecule function (MF) and nine cellular component (CC)), and four KEGG pathways (Fig. 5B) (Supplementary Table S15A in Additional file 1). As showed in Fig. 5A, the BP-GO terms were classified into clusters according to their pairwise similarities. The biggest cluster of BP-GO terms are involved in cell migration, locomotion, and related regulation, reflected through most significantly enriched BP-GO terms like positive regulation of cell migration (GO:0030335, FDR = 0.006), positive regulation of cell motility (GO:2000147, FDR = 0.006), and regulation of cell migration (GO:0030334, FDR = 0.006) among others (Supplementary Table S15A in Additional file 1). The second cluster of BP-GO terms are related to cell development, such as cell population proliferation (GO:0008283, FDR = 0.02), epithelial cell differentiation (GO:0030855, FDR = 0.01), and epithelium development (GO:0060429, FDR = 0.01), which are important for maintenance of mammary gland health. In addition, two BP-GO terms related to metabolic processes were enriched and clustered together, including lipid metabolic process (GO:0006629, FDR = 0.02) and small molecule biosynthetic process (GO:0044283, FDR = 0.02). Meanwhile, a KEGG pathway related to lipid metabolism, the fatty acid metabolism (bta01212, FDR = 0.03), was also enriched (Fig. 5B). This suggests that the hyper-methylated GE-dMHBs may participate in regulating mammary gland health and biological functions, such as cell migration, cell development, and metabolic processes, by mediating the repression of related genes during S. aureus subclinical mastitis. Furthermore, a total of 32 downregulated DEGs that negatively correlated with their overlapped hyper-methylated GE-dMHBs, such as ACTN1, CLDN3, CLDN4, CXCL17, CXADR, FGF1, FGF2, TGFB2, SPRY2, and SLC9A3R1, among others, were enriched in at least 10 GO terms and/or KEGG pathways related to the functions mentioned above, highlighting their important roles in mammary gland health (Supplementary Table S15B in Additional file 1).
Fig. 5
Functional enrichment for gene expression associated differential methylation haplotype blocks (GE-dMHBs). A, B Tree-plot showing the clustering of biological process GO terms (A) and enrichment map of four KEGG pathways (B), which were significantly enriched by downregulated differentially expressed genes (DEGs) that were negatively correlated with their overlapped hyper-methylated GE-dMHBs. C Enrichment map of 18 KEGG pathways significantly enriched by upregulated DEGs that negatively correlated with their overlapped hypo-methylated GE-dMHBs. Detailed data on the depicted findings are found in Supplementary Table 15
Besides, the 1075 hypo-methylated GE-dMHBs (negatively correlated with their overlapped 369 upregulated DEGs) were significantly enriched in 18 KEGG pathways, mostly immune and disease-related terms (Fig. 5C, Supplementary Table S15A in Additional file 1). For instance, some key functions of immune defense process were significantly enriched, including chemokine signaling pathway (bta04062, FDR = 0.002), cytokine-cytokine receptor interaction (bta04060, FDR = 0.002), leukocyte transendothelial migration (bta04670, FDR = 0.002), Th17 cell differentiation (bta04659, FDR = 0.004), natural killer cell-mediated cytotoxicity (bta04650, FDR = 0.011), and B cell receptor signaling pathway (bta04662, FDR = 0.047), among others (Fig. 5C). It is worth mentioning that 16 upregulated DEGs with hypo-methylated GE-dMHBs in their regulatory regions, including RAC2, IL1B, IL2RA, BOLA-DOB, SYK, and NCF1, among others, were enriched in five or more of the immune/disease-related KEGG pathways (Supplementary Table S15B in Additional file 1). These enriched KEGG pathways indicated that the absence or scarcity of DNA methylation in these GE-dMHBs may be a possible regulatory mechanism underlying the upregulated expression of related genes and potential regulation of the immune defense of mammary gland against S. aureus invasion. On the other hand, the GE-dMHBs that positively correlated (FDR < 0.05 and rho > 0.3) with their overlapped DEGs accounted for a small part (27%) of the GE-dMHBs and were enriched in few GO terms or KEGG pathways. Upregulated DEGs (n = 386) with correlated hyper-methylated GE-dMHBs (n = 1399) were enriched in three CC-GO terms and five KEGG pathways (Supplementary Table S15A in Additional file 1). Meanwhile, the 187 downregulated DEGs with correlated hypo-methylated dMHBs (n = 364) were enriched in four CC-GO terms only (Supplementary Table S15A in Additional file 1). Furthermore, we also submitted for functional enrichment the DEGs that correlated with their overlapped pro-, FE-, and FI-GE-dMHBs separately. In general, the functional annotations enriched by pro-, FE-, and FI-GE-dMHBs were similar with those of all GE-dMHBs (Supplementary Table S15C-E in Additional file 1, Fig. S10 in Additional file 2).
Identification of discriminant signatures from GE-dMHBsWe used DIABLO method to identify a subset of discriminant and highly correlated signatures across different omics by integrating the 6435 GE-dMHBs and their overlapped correlated DEGs (n = 1484). Firstly, the latent component of the two OMICs (GE-dMHBs and DEGs) were highly correlated (r = 0.90, Fig. 6A). After finetuning, five GE-dMHBs, including one pro-GE-dMHBs and four FI-GE-dMHBs, and seven DEGs discriminant signatures were found to explain the most variations delineating the SAP and HC cows (Fig. 6B–E, Supplementary Table S16 in Additional file 1). Although the discriminant GE-dMHBs were not directly located in the discriminant DEGs, they were significantly and strongly correlated with each other (Fig. 6B). The only pro-GE-dMHB, Chr29:46226206:46226215 located in the promoter of CPT1A, was the most important with highest loading weight (0.79). FI-GE-dMHB, Chr28:25477467:25477579 located in the first intron of SRGN, was the second most important (loading weight = 0.51). These two GE-dMHBs and the fourth discriminant signature, Chr3:105951347:105951407 located in the first intron of RLF, were all hypo-methylated in SAP group and strongly negatively correlated with the upregulated expression of their overlapped DEGs. The fifth signature, Chr22:17428396:17428497 located in the first intron of SRGAP3, showed positive correlation between its hypo-methylation and the downregulated expression of SRGAP3 in SAP group. While the third signature, Chr25:34469403:34469492 located in the fist intron of DTX2, was hyper-methylated in SAP group and positively correlated with the upregulated expression of DTX2. However, all seven DEGs were downregulated in SAP group. Among them, three (ACOT4, PDLIM5, and RAB11FIP5) were also selected as discriminant genes driving the major changes in gene expression profiles of key gene modules related to S. aureus subclinical mastitis in our previous transcriptome analysis [38].
Fig. 6
留言 (0)