
記住我


Pathologic angiogenesis is a hallmark of malignant tumors [1]. Growth of the primary tumor and metastatic progression depend on direct connection to the circulatory system for oxygen and nutrients [2]. Tumors can either co-opt existing blood vessels or create new ones via neoangiogenesis [3]. These new vessels, however, are often leaky, chaotic, and dysfunctional, contributing to the characteristic aberrant metabolic microenvironment defined by acidosis, hypoxia, and deranged glucose metabolism [4]. Yet, these defining dysregulated features also seem to drive a tumor’s ability to persist in and evade our otherwise hostile environment.
For over nearly half a century, targeting tumor angiogenesis to disrupt a tumor’s nutrient supply has been explored as an anti-tumor strategy [5]. Recently, interest in the mechanisms responsible for tumor-associated angiogenesis has been reinvigorated by the concept of normalizing the dysregulated vasculature of the tumor microenvironment (TME) as means to facilitate host immune response and influx of systemic therapies. The moderate success of anti-vascular endothelial growth factor (VEGF) targeting in metastatic colorectal cancer (CRC) has demonstrated the efficacy of anti-angiogenic therapies in real-world experience [6,7,8]. However, the realities of therapy resistance and limited applicability to a majority of solid organ malignancies make evident our nascent understanding of the mechanisms underlying tumor angiogenesis and how to target them [9].
C-type lectins are an evolutionarily conserved protein superfamily that boasts an extensive breadth of domain architecture, signaling pathways, and function [10]. Defined by their hallmark C-type lectin binding domain (a double-looped, two stranded antiparallel beta-sheet) aka a CTLD, this family of over 1000 mammalian members have been classified into 17 subfamilies, grouped by structural and functional similarities [11]. Group XIV proteins, including CD93 (C1qRp), thrombomodulin (CD141, TM), CD248/endosialin (tumor endothelial marker 1/TEM1), and C-type lectin family member 14a (CLEC14a), are a subfamily of transmembrane CTLDs with intimate involvement in physiologic angiogenesis, cell adhesion, and regulation of inflammation. Their importance to normal physiologic processes has been paradoxically highlighted by group XIV CTLDs’ emerging roles as significant drivers of tumor angiogenesis and metastatic dissemination [12, 13]. Further, preclinical studies demonstrate that the therapeutic targeting of group XIV CTLDs result in significant anti-tumor benefit, some currently being tested in the clinic.
Given the recent success in targeting the tumor vasculature via the VEGF pathway and the emerging role of CTLD proteins in cancer angiogenesis and progression [14,15,16], we aim to give a brief overview of the Group XIV CTLD members within the larger context of C-type lectins, review our current understanding of each group XIV protein’s role in normal biology as well as tumorigenesis, from a mechanistic and therapeutic perspective, provide a brief review of group XIV CTLDs in the context of other well-described lectins, and offer potential future direction in expanding our knowledge of the mechanisms behind CTLDs and the avenues to target them.
Group XIV C-type lectins: an overviewCTLDs are an evolutionarily conserved superfamily of proteins with vast variability in structure, binding ligands, and function [11]. The structural hallmark of CTLDs is a double-looped, two stranded antiparallel β-sheet binding site known as the C-type lectin binding domain. Significant sequence variability in CTLD structure in addition to unique domain architecture have enabled classification of 17 known subfamilies of mammalian CTLDs [17]. Originally, thought to strictly bind carbohydrates via a calcium dependent process, it is now known that many human CTLD members do not bind carbohydrates nor require calcium to bind its ligands including proteins, lipids, and inorganic molecules [17]. The diversity in binding ligands for this superfamily is mirrored by their diversity of function.
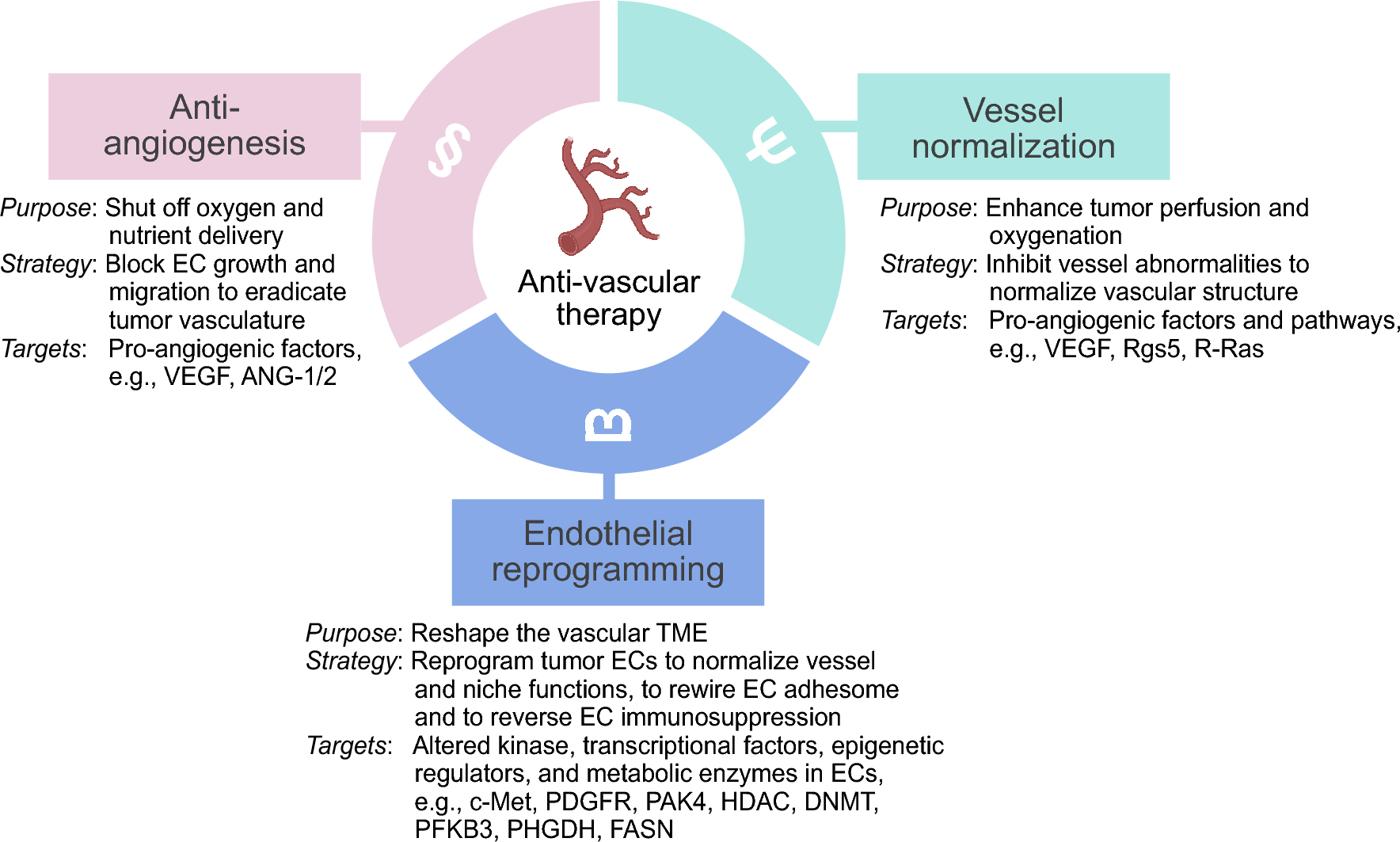
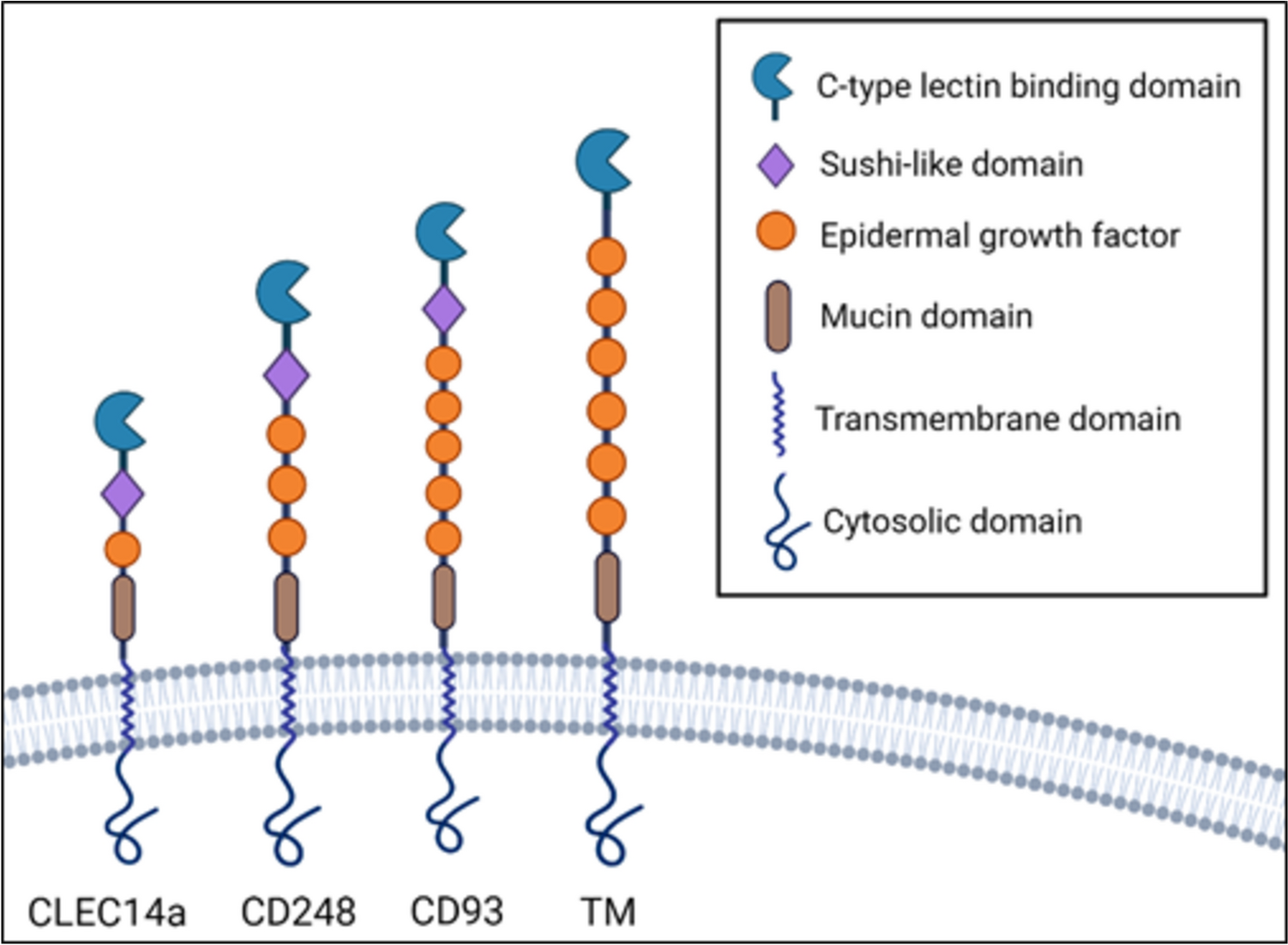
Group XIV subfamily of CTLD proteins are a relatively recent discovery among C-type lectins that share similar domain architecture, binding partners and expression, and function [18]. This family is composed of four members, including CD93, TM, CD248, and CLEC14a (Fig. 1). These proteins share a common architecture: an N-terminal signal peptide, CTLD containing conserved six cysteine residues (in addition to two non-canonical cysteines, except thrombomodulin which only has 4 conserved cysteine residues) followed by sushi domain (also known as complement control protein domain/short consensus repeat), a serine-proline-threonine rich region on the extracellular domain that allow for frequent modifications, namely glycosylation, a single pass transmembrane portion, followed by a cytoplasmic domain [19]. Each protein is distinguished by a variable number of epidermal growth factor (EGF) subunits. All of them can be membrane-bound or secreted in a soluble form, the latter of which often occurs via metalloproteases [20]. All group XIV proteins are preferentially expressed by mesoderm-derived cells such as endothelia and hematopoietic cells for TM, CD93, and CLEC14a, and mesenchymal cell-specific expression of CD248. Furthermore, shared binding partners amongst the group XIV proteins support their homology; CD248, CD93, and CLEC14a bind to endothelial extracellular matrix (ECM) protein multimerin-2 (MMRN2), although at different regions of their extracellular domains; TM and CD248 bind ECM glycoprotein fibronectin while TM and CLEC14a are known to both bind heat shock protein 70-1 (HSP70-1). [21, 22]
Fig. 1
Structural representation of group XIV C-type lectin domain proteins in transmembrane state. CLEC14a, C-type lectin 14a, TM thrombomodulin
In normal physiology, group XIV CTLDs have key roles in maintaining homeostasis by regulating vessel formation, ECM organization, and inflammatory responses. Thrombomodulin acts as a natural anticoagulant by binding to the serine protease thrombin which renders thrombin unable to convert fibrinogen to fibrin, ultimately prohibiting formation of a reactive platelet plug [23]. CD248 is unique in its expression on mesenchymal cells, particularly cells of blood vessel wall called pericytes that are responsible for vessel integrity [24]. CD93 and CLEC14a are proteins minimally expressed on endothelia but upregulated during intense angiogenesis of embryogenesis and in other pathologic conditions including inflammation and malignancy, facilitating vessel tube formation, endothelial adhesion and migration, and orchestrating surrounding stroma [25,26,27]. Taken together, the group XIV CTLDs, while slightly variable in structure and binding interactions, all appear to serve important roles in vasculogenesis.
Over the past thirty years, the role of group XIV CTLDs and C-type lectins at-large in cancer progression has risen to prominence. Various CTLD families such as selectins, dectins, and related lectins such as galectins are among the most well characterized glycoproteins harboring carbohydrate binding domains implicated in facilitating a pro-tumorigenic TME and metastasis, predominantly via recruitment and adhesion of various immune and hematopoietic cells, and even tumor cells themselves [28]. Among group XIV proteins, CD248, originally identified as a human fetal fibroblast antigen, was found to be the most upregulated endothelia-related gene in CRC tissue compared to normal tissue, thus garnering the designation, tumor endothelial marker 1 [29]. CD93 and CLEC14a were similarly among the most enriched genes comprising an angiogenesis signature of over 1000 tissue samples across three carcinomas [30]. The involvement of Group XIV proteins in tumor angiogenesis is without question, yet, the molecular mechanisms and implications for tumor progression and abrogation are still in their nascent phases. The following sections will review our current understanding of each group XIV protein’s known role in tumorigenesis, discuss therapeutic endeavors, and offer potential directions for future study.
CD93: overviewCD93 is a group XIV CTLD with five repeating EGF domains primarily expressed by endothelial cells (EC), neurons, and a mirage of myeloid cells such as macrophages, monocytes, and stem cells [18]. Previously referred to as complement component C1q receptor (C1qRp), it is now known that CD93 does not directly binding this complement protein but associates with various ECM proteins in the absence of calcium [19, 31]. In fetal mice, CD93 has been shown to be ubiquitously expressed on primitive hematopoietic cells, aorta, gonads, and mesonephros regions, with a predominant role in vascular remodeling given its specific expression on expanding branches of the aorta and neural plexuses on embryonic day 9–10. [32] Nonetheless, absence of CD93 during embryogenesis does not cause fatal consequences suggesting redundancy in its function. In adult humans and mice, CD93 expression wanes.
Thus far, facilitation of efferocytosis (removal of apoptotic cells), endothelial maturation, migration, and intercellular adhesion are proposed native functions of CD93. Rather than directly binding complement receptors, as once regarded, in vitro studies using transfected Chinese hamster ovary (CHO) cells, THP-1 macrophages, and primary human macrophage cultures, soluble CD93 has been shown to act as an opsonin to mediate macrophage recruitment and phagocytosis [33, 34]. As a cell-surface protein, CD93 appears to mediate endothelial function, namely adhesion with other ECs, migration, and interaction with the ECM, via interaction with various binding partners such as Multimerin-2 (MMRN2), β-dystroglycan, Cbl, and recently insulin growth factor binding protein 7 (IGFBP7) [14, 35, 36]. A series of studies from the University of Siena has elucidated much of our mechanistic understanding of CD93 interaction with MMRN2 and downstream pathways facilitating EC function. First, the importance of MMRN2 interaction with CD93 in promoting endothelial function was demonstrated by blocking CD93/MMRN2 interaction with a monoclonal antibody (mAb) against the CTLD of CD93 and resultant inhibition of human umbilical vein endothelial cell (HUVEC) adhesion, migration, and tube formation [35]. Next, elucidation of amino acid residues important for interaction between CD93 and MMRN2 via mutagenesis assays revealed proper folding of the CTLD and presence of a phenylalanine residue at position 238 to be crucial in binding MMRN2 and carrying out angiogenic roles [37]. Subsequently, the Siena group showed that CD93 interaction with β-dystroglycan, an ECM protein upregulated on activated ECs, is also important for endothelial activity via phosphorylation of CD93 extracellular domain, which in turn stimulates phosphorylation of downstream effectors such as Cbl [36]. Most recently, they demonstrated that CD93 preserves endothelial junctions via VE-cadherin interaction and suppression of phosphorylation via Rho signaling, thereby mitigating vessel permeability [38]. Recombinant forms of soluble CD93 comprised of only EGF repeats and the mucin domain of its CTLD have been shown to retain pro-angiogenic stimulation on ECs in vivo with perhaps more potent angiostimulation via the EGF domains [39]. Together, this data clearly defines a role for CD93 in mediating angiogenic processes and requires specific ligands and environments, such as malignancy, to execute its function.
CD93: role in malignancyWhile CD93 is preferentially expressed on ECs, they are at a minimal or absent level in normal human and mouse adult tissue. Recent studies have shown CD93 to be intensely upregulated on tumor-associated endothelium, supported by its status as one of the top 20 genes associated with human primary tumor angiogenesis signature surveying head and neck, breast, kidney, and brain tumors [30]. In subsequent studies, CD93 has been shown to be a downstream effector of VEGF, a potent stimulator for tumor-derived angiogenesis, and its expression significantly downregulated upon VEGF inhibition [14]. From single cell gene expression to surface protein expression, CD93 expression is by-far predominantly upregulated on solid organ malignancies and, to a lesser extent, some hematologic malignancies. In a human pan-cancer analysis of transcriptomic and mutational data from several online databases, CD93 was found to be upregulated in most cancers such as cholangiocarcinoma, ovarian, pancreatic, gastric, melanoma, and kidney but down regulated in lung adenocarcinoma and bladder urothelial cancer [40]. Prognostically, higher CD93 expression levels were correlated with worse oncologic outcomes in kidney renal papillary carcinoma, glioma, ovarian cancer, and uveal melanoma. In this same CD93-specific screening investigation, increased CD93 expression correlated with higher number of infiltrating T cells in some cancers but also higher immunosuppressive macrophage presence [40]. Immune-related genes known to promote cancer growth, tumor angiogenesis, and tumor metastasis were also positively correlated with CD93 upregulation. CD93 expression and upregulation on tumor endothelium has also been confirmed with immunostaining. Langenkamp et al. demonstrated that among low to high grade gliomas, higher grade gliomas had tumor vasculature with higher degree of CD93 positivity compared to lower grade gliomas [41]. In another study of human gliomas, Ma et al. showed CD93 expression was positively correlated with infiltration of immunosuppressive immune cells such as immunosuppressive macrophages, T regulatory cells, and myeloid derived suppressor cells (MDSCs) [42]. Similar cell-surface staining has been shown in carcinomas of the kidney, colon, and pancreas, supporting the notion that CD93 is upregulated on blood vessels supporting several types of tumors. [14]
In vitro and in vivo preclinical models exploring the roles of CD93 in angiogenesis, tumor progression and metastases build upon the foundation of our current mechanistic understanding of CD93. In in vitro studies of human dermal microvascular endovascular cells (HDMECs) treated with VEGF, CD93 knockdown cells failed to engage in cell adhesion and form tubes compared to untreated cells but rescued with lentivirus transfection of wild type (WT) CD93 with native cytoplasmic region [38]. Further, CD93 expressing ECs became disorganized and lost adhesion properties upon CD93 knockdown. In preclinical in vivo models, Langenkamp et al. demonstrated CD93 KO (KO) mice with orthotopically implanted mouse gliomas experienced longer survival and slower tumor growth compared to control mice [41]. Further, in subcutaneous fibrosarcoma model, CD93 KO mice had reduced tumor size. In histologic and immunofluorescent analyses, gliomas in CD93 KO mice were infiltrated by endothelia with abnormally polarized lumens and increased vascular permeability compared to controls, suggesting a high degree of vessel dysfunction in the absence of CD93.
Ligand interaction with CD93 appears to mediate its role in physiologic endothelial activity as well as tumor angiogenesis. Analysis of The Cancer Genome Atlas and endothelial specific online database (EndoDB) demonstrate that IGFBP7, a secreted ECM protein implicated in a variety of homeostatic processes, is upregulated on tumor endothelium [43]. Our lab has recently demonstrated specificity of IGFBP7 binding to CD93 [14]. Out of a library of over 6000 surveyed cell surface and soluble proteins, not including MMRN2, IGFBP7 was the only positive binding partner with CD93. The knockdown of IGFBP7 in HUVECs prevented tube formation compared to WT controls. Further, the importance of CD93 binding to IGFBP7 for angiogenesis was demonstrated by knockdown of CD93 in HUVECs and the absence of tube formation and EC migration with the addition of IGFBP7 protein but restored function with the addition of IGFBP7 in WT HUVECs. Likewise, the role of CD93-IGFBP7 interaction in tumor angiogenesis seems to be important. Blockade of the interaction with anti-IGFBP7 antibody similarly reduced tumor growth while promoting tumor vessel maturation, as evidenced by increased pericyte coverage and reduction in integrin β1/CD29 activation, a marker of EC destabilization and vascular leakiness. Similarly, Xu et al. implanted murine melanoma tumors with and without transfected IGFBP7 and found that IGFBP7 tumors had significantly more intratumoral vessels on immunostaining [44]. Furthermore, expression of IGFBP7 mutants that lose CD93 binding in tumors did not increase vessel densities as seen in tumors expressing WT IGFBP7, implicating a role of IGFBP7 in upregulating tumor angiogenesis via CD93.
CD93: therapeutic approachesRecent work detailing blockade with a CD93 mAb has led to interesting and promising results toward tumor vessel normalization rather than depletion as seen in antibody-directed binding to other group XIV CTLDs such as CD248 and CLEC14a as will be discussed in subsequent sections (Fig. 2a). CD93 signaling inhibition with anti-CD93 mAb led to tumor vessel normalization rather than depletion as evidenced by maturation of blood vessels with increased pericyte coverage via upregulated immunofluorescent staining of alpha-smooth muscle actin (αSMA) and neural/glial antigen 2 (NG2) coverage of vessel structures [14]. Additionally, blockade of CD93 resulted in improved perfusion of subcutaneously implanted pancreatic and melanoma tumors, suggested by decrease in intratumoral hypoxia with improved tomato lectin staining of intratumoral vessels and hypoxic inducible factor 1 alpha (HIF1α) expression—this is in direct opposition to results reported in CD93 KO gliomas where CD93 knockdown mice exhibited worse perfusion than WT counterparts [41]. The vessel maturation induced by CD93 blockade resulted in exciting downstream therapeutic benefits—subcutaneous melanoma and pancreatic tumors had decreased growth, improved efficacy of chemotherapy delivery, and increased influx in anti-tumor immune cells such as CD8 + T cells and natural killer cells synergizing the effect of immunotherapy. However, the effects of CD93 blockade on orthotopic tumors, known to display tumor microenvironments more similar to human cancers, is a subject of ongoing investigation. Nonetheless, these promising preclinical results have led to initiation of a phase one clinical trial (Study DC-6001-101) (Table 1). [45]
Fig. 2
Examples of anti-tumor therapeutic approaches targeting Group XIV CTLDs. a Monoclonal antibody (mAb) specifically targeting CTLD with either cellular depleting effects via antibody-dependent cell mediated cytotoxicity (ADCC) or blocking function inhibiting downstream signaling b Antibody drug conjugate (ADC) targeting CTLD to induce receptor mediated endocytosis of cytotoxic conjugate (CC) in form of DNA intercalators or inhibitors of cell division resulting in cell death c Anti-cancer vaccination with immunogen such as fused DNA construct of CTLD and tetanus toxoid stimulating CTLD specific cytotoxic T lymphocytes (CTLs) targeting malignant CTLD-expressing cells d Chimeric antigen receptor T (CAR T) cell therapy engineered with high specificity antibody binding fragments (VH & VL) targeting CTLD and intracellular T cell stimulating motifs to activate cytotoxicity via cytokines (interferon gamma). VEGF/R, vascular endothelial growth factor/receptor
Table 1 Summary of group XIV CTLDs’ characteristicsCD93 has been demonstrated to play a significant role in vessel formation, both by normal and pathologic processes. Its upregulation in a plethora of solid organ tumors, and promising anti-tumor effects when disrupted, especially given its distinct vascular normalization effects, give credence to further investigation into how CD93 acts as an intercellular adhesion molecule and promoter of EC formation. Further, the expression of CD93 on EC of the lung and liver gives impetus to investigate the normal physiologic function of CD93 in these organs and how this function is dysregulated, if at all, in metastatic dissemination.
CD248: overviewCD248, also named endosialin due to its high degree of glycosylation, shares the common structure with its fellow group XIV CTLDs with the exception of containing three EGF repeat domains [26]. Like CD93, CD248 is absent (brain, stomach, skin, ovary) or minimally expressed (small intestine, uterus, kidney) on adult normal tissue but pronounced during fetal development, yet deficiencies in CD248 expression during this stage seem to be produce no postnatal effects [46]. CD248 is unique from its group XIV kin in that it is not expressed on ECs, contrary to its previous name tumor endothelial marker 1, but only mesenchymal cells including stromal fibroblasts, smooth muscle cells, pericytes, mesenchymal stem cells, and naïve T cells [47, 48]. Its role during embryogenesis involves vascular remodeling and angiogenesis. In vitro studies suggest CD248 remodels developing vasculature via vessel pruning and branch regression [49]. Further, the silencing of CD248 in cultured human fibroblasts leads to reduced capacity to migrate and proliferate. CD248 has also been shown to have a role in promoting inflammation—in autoimmune diseases, CD248 is known to be upregulated on fibroblasts and pericytes of synovial tissue and mesenchymal cells of the skin as KO of CD248 cytoplasmic domain showed significant reductions in inflammatory cytokines, synovitis, and arthritis in murine models. [19, 50]
Mechanistically, CD248 is proposed to exist within a complex interplay involving various ECM proteins and signaling pathways mediating stromal cell migration, activation, and proliferation. CD248 has been shown to bind to ECM proteins collagen I, collagen IV, and fibronectin [51]. In a series of elegant in vitro studies, Tomkowicz et al. demonstrated that CD248 expression level correlated with fibronectin expression, and that CD248 expression facilitated adhesion of cells to fibronectin, resulting in unique, web-like morphology and enhanced migratory capacity of CD248 expressing CHO cells. Furthermore, production and secretion of matrix metalloprotease-9 (MMP-9), a known enzymatic protein responsible for basement membrane remodeling, was found to be upregulated in the supernatant of CHO expressing CD248 cells, which the authors hypothesized could contribute to the enhanced migratory phenotype of CD248 CHO cells in vitro. Importantly, the authors also developed a humanized IgG mAb to CD248 (MORAb-004, commercially ontuxizumab) that blocked CD248 binding to its ECM ligands. MORAb-004, as discussed in subsequent sections, demonstrated the validity of CD248 targeting in preclinical tumor models, and has advanced to clinical trials.
The cytoplasmic domain of CD248 seems to facilitate angiogenesis-independent stromal activity. Maia et al. demonstrated that the presence of CD248 cytoplasmic domain suppresses expression of tumor suppressor transgelin (SM22a), a repressor of transforming growth factor-beta (TGF-β) and MMP-9 expression, thus the absence of the cytoplasmic domain upregulated SM22a and decreased MMP-9 activity [22]. Furthermore, in a transwell migration assay, WT fibroblasts expressing CD248 migrated much further than CD248 cytoplasmic domain null counterparts in a platelet-derived growth factor-BB (PDGF-BB) dependent manner, suggesting migration is facilitated by the cytoplasmic domain. Lastly, tumor cell viability may be affected by loss of CD248 cytoplasmic domain as coculture of WT and cytoplasmic domain-absent CD248 with T241 fibrosarcoma cells resulted in reduced tumor cell viability after 48 h. In sum, CD248 uses a variety of mechanisms to facilitate stromal remodeling, many of which are dysregulated in pathologic conditions.
Interestingly, CD248 has also been shown to be expressed on a specific subset of human naïve CD8 T cells in the thymus and peripheral blood. The definitive role of CD248 on T cells has yet to be elucidated, however Hardie et al. has begun to explore the anti-proliferative function of endosialin in vitro [48]. They speculate that the decrease in CD248 expression in extrafollicular zones of secondary lymphoid structures retains CD8 + T cells in antigen recognition areas, promoting a quiescent T cell state. However, the anti-proliferative role of CD248 on CD8 + T cells has not been demonstrated given the lack of expression on murine T cells nor is it known if tumor associated CD8 + T cells express CD248 differentially than other effector T cells.
CD248: role in malignancyCD248 was originally deemed TEM1 given its status as the highest upregulated gene in a seminal serial analysis of gene expression (SAGE) of endothelial genes found in malignant CRC samples compared to normal tissue [
留言 (0)