
記住我
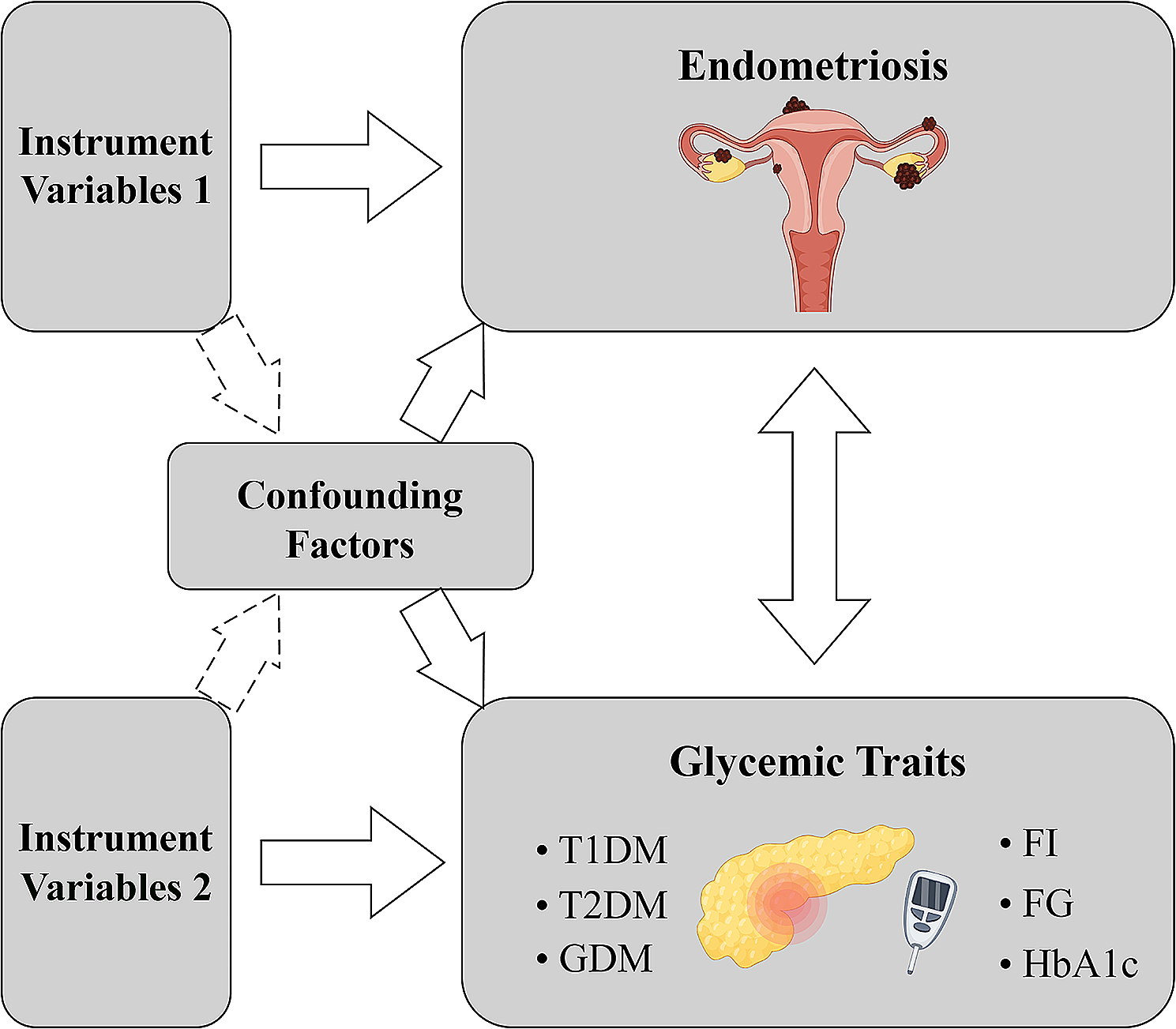
The flow chart of this study is shown in Fig. 1. The patients were grouped according to diabetes status, and the differences in clinical characteristics among the non-diabetes group, pre-diabetes group and type 2 diabetes group were compared. The results showed that gender, age, race, education level, BMI, TC, TG, HDL, LDL, FPG, Fins, Hba1c, drinking history, smoking status, and history of hypertension significantly differed among the three groups (Table 1). Although the incidence of lung cancer was low in the study cohort, the incidence of lung cancer gradually increased among the three groups.
The results of whether or not lung cancer occurred are shown in Fig. 2; FPG and glycosylated haemoglobin were significantly different between the two groups. FPG and glycated haemoglobin levels (Fig. 2A and B) were higher among participants with lung cancer. Although the fasting insulin level was lower in the lung cancer group than in the non-lung cancer group, the difference between the two groups was insignificant (Fig. 2C).
Fig. 2
The Association between diabetes and lung cancer. (A-C) FPG, glycosylated haemoglobin, and fasting insulin in the lung cancer group and control group; (D) The association between FPG and the risk of lung cancer; (E) The association between glycosylated haemoglobin and the risk of lung cancer
The association between diabetes status and lung cancerTable 2 shows the association between diabetic status (non-diabetic, pre-diabetic, and type 2 diabetes) and lung cancer using multiple logistic regression analysis. The results were presented with odds ratios (ORs) and 95% confidence intervals (CIs) of the three different models. The ORs of all three models were considered the reference group for the non-diabetic group. For the pre-diabetes group, the OR value of model 1 was 6.019 (95%CI 2.317, 15.637, p = 0.00023), and the OR value of model 2 was 3.334 (95%CI 1.259, 8.829, p = 0.01540). The OR value of model 3 was 3.289 (95%CI 1.231, 8.788, p = 0.01760). For the type 2 diabetes group, the OR of model 1 was 7.033 (95%CI 2.441, 20.259, p = 0.00030), and the OR of model 2 was 3.032 (95%CI 1.015, 9.054, p = 0.04689). The OR of model 3 was 3.110 (95%CI 0.999, 9.684, p = 0.05020).
Table 2 Relationship between diabetes status and lung cancerThese findings suggest that T2DM is significantly associated with increased risk for lung cancer, even after adjustment for multiple covariates. The results are detailed in Table 2.
In U.S. adults, fasting blood glucose and glycosylated haemoglobin were nonlinearly associated with lung cancer risk. As shown in Fig. 2, fasting blood glucose and glycosylated haemoglobin had an inverted U-shaped relationship with the risk of lung cancer (P for non-linear < 0.001). In summary, diabetic status and glycemic measures were significantly associated with an increased risk of lung cancer.
Identification of differentially expressed genes (DEGs)This study analyzed datasets obtained from the GEO database using R language. The lung cancer group was compared with the control group, and 4741 DEGs were found, including 2358 up-regulated genes and 2383 down-regulated genes. A total of 334 DEGs (including 92 up-regulated genes and 242 down-regulated genes) were identified between T2DM patients and normal controls. Subsequently, the intersection of NSCLC-DEGs and T2DM-DEGs was taken on the R platform to analyze their co-DEGs, and the results were visualized using a Venn diagram (Fig. 3C). We identified 57 co-DEGs, including 25 up-regulated co-DEGs (RACGAP1, MYBL2, ASUN, SMC4, HN1L, BRI3BP, ORC5, UNG, SMC6, ALG6, DROSHA, GOLT1B, FAM69A, G2E3, ABCE1, NCAPD3, RNMT, CDC27, YWHAG, SLC5A3, DLG1, POT1, CDC7, PIGW, ZNF322) and 32 down-regulated co-DEGs(NXB, GSTM5, RILPL2, PREX1, HSPB8, AGTR1, ARRB2, NCF2, FAXDC2, SELPLG, ALDH2, NCF1C, CFP, NCF4, IL6R, ARID5A, NCF1, IL16, MID1IP1, PALM, MCL1, ATP6V0D1, TFEB, FRAT1, SIGLEC5, RAB24, LSP1, PYCARD, SOD2, TREM2, CAPG, ADGRG3). Figure 3A-B shows the volcano diagram.
Fig. 3
volcano maps of (A) Lung cancer and (B) T2DM;(C) Venn diagram of co-DEGs
Identification of co-expression modules by WGCNAWGCNA was used to construct a co-expression module to evaluate whether there was a co-expression pattern of each gene between samples and to determine whether NSCLC genes and T2DM genes had the same expression pattern in a particular stage. By constructing a weighted gene co-expression network, we set the soft threshold to 12 to guarantee high gene independence and low average connectivity to identify co-expressed gene modules. Based on the weighted correlation, hierarchical clustering analysis was conducted, and the cluster results were determined according to the set criteria. The analysis outcomes were depicted using cluster trees with different branches and colors. In this study, we analyzed the expression matrices of all samples in the NSCLC and T2DM datasets separately. We selected variant genes in the top 30 to 50% (less than 5000) for co-expression analysis. We calculated module signature genes representing each module’s overall gene expression level clustered according to their correlation. In addition, we generated the heatmap to show the correlation between modules and a given trait or grouping, with the trait or grouping on the abscordinate and the module on the ordinate; the redder the color in the heatmap, the stronger the positive correlation. On the contrary, the bluer the color, the stronger the negative correlation. The values in the grid are the correlation coefficients and p-values, respectively. If a trait or grouping is linked to a module with an absolute value closer to one, it is likely associated with the trait or grouping’s gene function in that module. WGCNA identified eight modules in the lung cancer data, and the interrelationship between the modules was assessed. MEmagenta exhibited a highly negative correlation with NSCLC, while MEgreen showed a positive correlation with NSCLC. In addition, 17 modules were found in the diabetes data, with the MEorangered4 module displaying a negative correlation with T2DM, and the MElightcyan1 module was highly positively correlated with T2DM. We retained the genes associated with these modules for further analysis. The results of all WGCNA analyses are shown in Fig. 4.
Fig. 4
WGCNA for (A,D) the scale-free index for various soft-threshold powers (β) and the mean connectivity for various soft-threshold powers for NSCLC and T2DM; (B,E) phyloclustering tree map of the genes for NSCLC and T2DM; (C,F) heatmap of module and trait / grouping correlation for NSCLC and T2DM
Functional enrichment analysisGO and KEGG enrichment are two main methods for analyzing gene function and structure. Firstly, GO and KEGG enrichment analyses were performed on lung cancer. GO enrichment analysis identified the biological process (BP), cellular component (CC), and molecular function (MF) of these genes, respectively. From the GO analysis of NSCLC (Fig. 5A), biological process (BP) (Fig. 5B) showed that DEGs were mainly enriched in an extracellular matrix organization, extracellular structure organization, extracellular matrix organization, mitotic nuclear division, cell chemotaxis, and mitotic sister chromatid segregation. The cellular component (CC) (Fig. 5C) consisted mainly of collagen-containing extracellular matrix, condensed chromosome-centromeric region, chromosome-centromeric region, and condensed chromosome kinetochore, kinetochore. Molecular functions (MF) (Fig. 5D) mainly included peptidase regulator activity, glycosaminoglycan binding, and enzyme inhibitor activity. KEGG pathway analysis (Fig. 5E-F) showed that DEGs were mainly enriched in the Cell cycle (p = 0.00011), Complement and coagulation cascades (p < 0.0001), Staphylococcus aureus infection (p = 0.00068), Hematopoietic cell lineage (p = 0.00168), Cell adhesion molecules (p = 0.002091), Viral protein interaction with cytokine and cytokine receptor (p = 0.000448), Antifolate resistance (p = 0.008433), and p53 signaling pathway (p = 0.028114).
GO analysis of T2DM-DEGs is shown in Fig. 6A. Biological processes (BP) (Fig. 6B) showed that DEGs were mainly enriched in the regulation of intrinsic apoptotic signaling pathways, regulation of apoptotic signaling pathways, and superoxide metabolism. The cellular component (CC) (Fig. 6C) consists mainly of NADPH oxidase complexes, Fleming bodies, endocytic vesicles, and secondary lysosomes. Molecular function (MF) (Fig. 6D) mainly includes NADH oxidase activity for superoxide production, oxidoreductase activity, NADPH oxidase activator activity for superoxide production, and DNA-glycosylase activity. KEGG pathway analysis (Fig. 6E-F) was mainly enriched in several metabolic diseases, such as Lipid and atherosclerosis (p = 0.003051), fat digestion and absorption (p < 0.0001), and some signaling pathways, such as neurotrophic factor signaling pathway (p = 0.022672)and AGE-RAGE signaling pathway (p < 0.0001).
In addition, GO functional enrichment analysis and KEGG enrichment analysis were performed to explore co-DEGs’ biological functions further. GO pathway analysis of co-DEGs (Fig. 7A) showed that Changes in biological processes (BP) (Fig. 7B) mainly include the superoxide metabolic process, reactive oxygen species metabolic process, and reactive oxygen species metabolic process. The cellular component (CC) (Fig. 7C) was mainly enriched in NADPH oxidase complex, secondary lysosome, and Flemming body. In terms of molecular function (MF) (Fig. 7D), co-DEGs were mainly enriched in superoxide generating NADPH oxidase activator activity, superoxide generating NAD(P)H oxidase activity oxidoreductase activity, acting on NAD(P)H, and oxygen as acceptor. As for the enrichment of KEGG analysis (Fig. 7E-F), the result of the co-DEGs mainly enriched in some inflammation and metabolic diseases, such as Lipid and atherosclerosis (p = 0.001190)), Neutrophil extracellular trap formation (p = 0.005241), Diabetic cardiomyopathy (p = 0.006495), Leukocyte transendothelial migration (p = 0.008752), and Chemical carcinoma-reactive oxygen species (p = 0.009001), and expressed in signaling pathways, such as Chemokine signaling pathway (p = 0.033944) and PI3K-Akt signaling pathway (p = 0.042931), and some cellular life processes, Examples include Osteoclast differentiation (p = 0.001493), Phagosome (p = 0.002308), and Cell cycle(p = 0.002596).
Fig. 5
Functional characteristics analysis for NSCLC. (A) GO enrichment results. (B) Go-enriched BP; (C) Go-enriched CC; (D) GO MF; (E) KEGG enriched barplot; (F) Dot plot of KEGG enrichment
Fig. 6
Functional characteristics analysis for the T2DM. (A) GO enrichment results. (B) Go-enriched BP; (C) Go-enriched CC; (D) GO MF; (E) KEGG enriched barplot; (F) Dot plot of KEGG enrichment
Fig. 7
Functional characteristics analysis for the co-DEGs. (A) GO enrichment results. (B) Go-enriched BP; (C) Go-enriched CC; (D) GO MF; (E) KEGG enriched barplot; (F) Dot plot of KEGG enrichment
Construction of PPI network and screening of key genesIn this study, the STRING database was used to construct the PPI network of up-regulated and down-regulated co-DEGs to screen the hub genes of co-DEGs further. The PPI network of co-DEGs that were both up-regulated consisted of 21 genes and 296 edges, and the PPI network of co-DEGs that were down-regulated consisted of 29 genes and 448 edges (Fig. 8A-B). Key hub genes were screened using the cytoHubba plugin in Cytoscape software. According to the visualization results of the PPI network combined with the key nodes of the PPI network, the hub key genes of 10 co-DEGs were finally screened out in this study, which were SMC6, CDC27, CDC7, RACGAP1, SMC4, NCF4, NCF1, NCF2, SELPLG and CFP (Fig. 8C-D).
Fig. 8
Protein-protein interaction (PPI) analysis of (A) upregulated co-DEGs; (B) downregulated co-DEGs; (C) hub genes in downregulated co-DEGs; (D) hub genes in upregulated co-DEGs
Receiver operating characteristic (ROC) curveROC curve was used to verify the diagnostic value of hub genes in NSCLC and T2DM. For patients with NSCLC, the AUCs of SMC6, CDC27, CDC7, RACGAP1, SMC4, NCF4, NCF1, NCF2, SELPLG, and CFP were 0.920, 0.920, 0.812, 0.968, 0.974, 0.920, 0.875, 0.974, 0.939, and 0.935, respectively (Fig. 9A-J). For patients with T2DM, the AUCs of SMC6, CDC27, CDC7, RACGAP1, SMC4, NCF4, NCF1, NCF2, SELPLG, and CFP were 0.812, 0.912, 0.901, 0.857, 0.801, 0.864, 0.831, 0.794, 0.846, and 0.860, respectively (Fig. 10A-J).
The hub-genes had good diagnostic efficiency and high diagnostic value in both NSCLC and T2DM (0.9 > AUC > 0.7). In addition, among the up-regulated genes, RACGAP1 had a high diagnostic value for NSCLC and T2DM. SMC4 had a high diagnostic value for NSCLC and T2DM among the down-regulated genes.
Fig. 9
ROC curve of co-DEGs in NSCLC. (A) SMC6; (B) CDC27; (C) CDC7; (D) RACGAP1; (E) SMC4; (F) NCF4; (G) NCF1; (H) NCF2; (I) SELPLG; (J) CFP
Fig. 10
ROC curve of co-DEGs in T2DM. (A) SMC6; (B) CDC27; (C) CDC7; (D) RACGAP1; (E) SMC4; (F) NCF4; (G) NCF1; (H) NCF2; (I) SELPLG; (J) CFP
Evaluation of immune cell infiltrationBox plots of differences in immune cell infiltration showed that compared with the control group, memory B cells, activated myeloid dendritic cells, M0 and M1 macrophages, plasma cells, CD4 + memory activated T cells were significantly increased in the lung cancer group. However, resting myeloid dendritic cells, eosinophils, activated mast cells, Monocytes, neutrophils, and CD8 + T cells were significantly reduced in the lung cancer group (Fig. 11A). The corrplot package in R software was used for the correlation analysis of immune cells. As shown in Fig. 11B, the numbers in the squares represent the correlation coefficients between the corresponding immune cells. The combinations with high positive correlation include memory B and plasma cells, Eosinophils and Monocytes. The combinations with high negative correlation were Eosinophils and plasma cells.
Then, we further explored the spear-man correlation coefficient between hub genes and the degree of infiltration of immune cells. As a result, all hub genes were associated with immune cells. Using correlation scatter plots, we visualized the six hub genes most strongly associated with immune cells (Fig. 12). NCF1, 2, 4 and SELPLG genes were positively correlated with B cells, CD4 + T cells, macrophages, neutrophils, and dendritic cells. The CFP gene was positively correlated with CD4 + T cells, neutrophils, and dendritic cells.
Fig. 11
(A) Analysis of differences in immune cells between the lung cancer and the control group. The horizontal axis represents the different immune cells, the vertical axis represents the proportion of immune cells; (B) Immune cell proportion correlation matrix. *p < 0.05;**p < 0.01;***p < 0.001;****p < 0.0001
Fig. 12
Correlation between hub genes and immune cell components in NSCLC and T2DM. (A) CDC27; (B) NCF4; (C) NCF1; (D) NCF2; (E) SELPLG; (F) CFP
留言 (0)