We describe the clinical manifestations observed in 5 patients with homozygous novel likely disease-causing variants in the MADD gene that presented with neurodevelopmental delay as well as arthrogryposis. Considering the previously reported patients, a variable neurodevelopmental delay was observed ranging from remarkable global developmental delay with early lethality to mild intellectual disability and longer survival [3, 4]. Four of our patients presented with the severe end of the spectrum while one showed the milder presentation. Hypotonia is a cardinal feature of MADD gene dysfunction being reported previously in 100% of cases in the lethal group [3, 4]. Likewise, failure to thrive (>70% of cases), hematological abnormalities (>90% of cases), and neonatal hypoglycemia (>90% of cases) are consistent features in the lethal group [3, 4]. Microcephaly (<−2 SD) was documented in 26% of the previously reported patients [3, 4] and 14% of our patients (one case). In contrast, the initial report showed relative macrocephaly in one patient [2]. Based on our results and those reported before [3, 4], the deceleration in the weight and length are more significant compared to the head circumference, thus giving the impression of relative macrocephaly. Endocrine dysfunction was reported in 80% of patients of the lethal group [3] but none of our patients. Given the early lethality, it is possible that some phenotypic traits are underreported in the existing literature. Frequencies of clinical features reported to date among patients with MADD variants compared with our cohort are provided in Table 1.
Arthrogryposis appears likely to represent an important feature of the condition. Notably, it was observed in all our patients (100%) and in 41% of those reported previously although provided no further details [3, 4]. It is typically congenital and shows a distal pattern of hands and feet although contracture of knee and elbow was observed in 2 patients (2/14; 14%) in a previous report [3]. Taken together with our present findings, these data provide compelling evidence for the association of arthrogryposis with MADD deficiency. Further reports would help to substantiate this observation. Therefore, we suggest that arthrogryposis should be added as one of the characteristic features for MADD-related neurodevelopmental disorder.
Patient 5 showed mild intellectual disability and myopathy whereas Patient 4 had a high CPK level. In contrast, Patient 1 in our cohort showed normal electromyogram (EMG) and nerve conduction studies. The defective neuromuscular transmission was seen in Madd-deficient mice [6]. However, the interaction of MADD with KIF1A that is a motor protein mediating the axonal transport of Rad3-positive vesicles [9] and the reduced pain sensations among patients of group 1 of Schneeberger and co-authors [3] gave evidences of hereditary sensory and autonomic neuropathy type II features among patients of this group. These results seem challenging. We recommend EMG and nerve conduction studies as well as the CPK measurement in any case with MADD deficiency. Although heterozygous missense variants in MADD have been also associated with muscular dystrophy [10], but the heterozygous parents (of our Patients 3 and 4 who had missense variant) were unaffected.
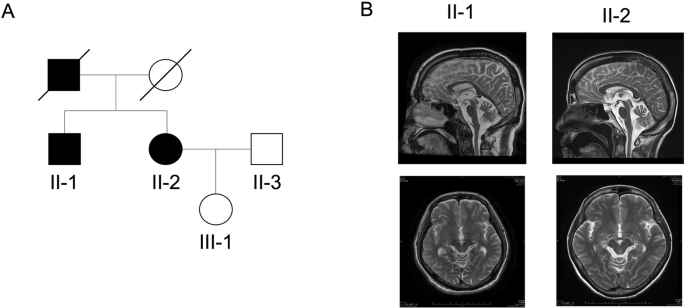
Brain MRI showed normal results in most of the reported cases (19/27; 70%). However, increased intra-and extra-axial CSF spaces were reported in 8 patients (8/27; 30%) and two also showed mega cisterna magna [3, 4]. Moreover, a hypoplastic pituitary was found previously in 2 patients; (2/27; ~7%) and was considered one of the main risk factors of hormone deficiencies [3]. The increased intra- and extra-axial CSF spaces are either indicative of brain atrophy or an increased production of cerebrospinal fluid in these patients. The cerebrospinal fluid has an important dynamic role in the central nervous development system [11]. In this study, we also noted asymmetric dilatation of lateral ventricles, hypoplastic corpus callosum and under-opercularization in four patients. These non-specific features argue against their clinical utility. Unexpectedly, Patient 1 in this study had brainstem and cerebellar hypoplasia. Reports of additional cases are needed to confirm the association between these brain imaging findings and MADD dysfunction.
Genital anomalies (micropenis and undescended testis) have been previously reported in 70% (12/17) of male patients with MADD variants and also in one of our male patients (33%). Thus, they appear likely to represent an important feature of the condition [3, 4].
Interestingly, congenital heart disease was not described as a consistent feature, reported in 2/14 (14%) of cases [3]. The cases we describe and those described by Abu-Libdeh et al. [4], have a much more prevalence of congenital heart disease showing 80% and 100%, respectively. It may be related to the periodic apnea described in these patients as 30% of cases showed variability in heart rate [3] but the pathogenesis of the periodic apnea is still unclear. Therefore, we propose that regular cardiac monitoring is performed.
Generalized tonic clonic seizures occurred in more than half of our patients and those reported in the literature [3, 4] and typically developed in the neonatal period and showed good response to antiepileptic drugs. The febrile illness and/or hypoglycemia-triggered seizures (usually presenting in the first year of life) are evident in 35% (5/14) of reported patients [3].
In total, 25 different MADD variants have been reported in 34 individuals [1,2,3,4]. They include 11 missense, 6 nonsense, 3 frameshfit, 4 splice, and one large deletion (encompassing exons 11 to 24). Variants were scattered across the coding region with no hot-spot exons. In this study, we identified three new MADD variants including two protein truncating [c.2620 C > T (p.Arg874 Ter) and c.4321delC (p.Gln1441ArgfsTer46)] and one missense [c.4307 G > A (p.Arg1436Gln)]. The two protein truncating variants are predicted to result in nonsense-mediated mRNA decay. On the other hand, the missense variant p.Arg1436Gln affects a highly conserved amino acid residue and was deemed deleterious by multiple bioinformatic tools. In addition, it was not found in our in-house database of >1500 exomes of Egyptian origin. Identification of more patients with this variant and future functional studies are warranted. This would help to confirm its pathogenicity. As such, we raise the total number of MADD disease-causing variants to 28 (Supplementary Table 4).
Among the reported variants, only four were recurrent including the p.Arg327* which was reported in 5 unrelated families, in the homozygous form in one family [3] and in the heterozygous state along with other variants in 4 families [1, 3]. Interestingly, all patients carrying the p.Arg327* were of European origin (Supplementary Table 3). The p.Arg327* is present in the heterozygous state in 47 normal individuals (MAF = 0.000029) according to gnomAD version 4, 45 of them were Europeans. Therefore, it is common and may be a founder MADD variant among Europeans. Another founder variant (c.2816+1 G > A) was described in 7 patients from 4 unrelated Arab-Muslim families [4]. Additionally, the p.Pro354Leu and p.Ser1213* variants were reported in the heterozygous state twice in two unrelated Europeans families each [1, 3]. Other variants are unique and appear to be private as each found in one family.
Our results support the findings of Schneeberger and co-authors [3] and confirm the absence of phenotype-genotype correlations in patients with MADD variants. Missense variants were identified among many patients of the severe end [3]. Likewise, Patients 3 and 4 in our study harboring the missense variant c.4307 G > A (p.Arg1436Gln) presented with a severe neurological phenotype, multiple organ dysfunction and showed early lethality. In comparison, Patient 5 whom carried a homozygous nonsense variant in exon 15 displayed a relatively milder phenotype and longer survival.
In conclusion, our report confirms the core phenotype associated with biallelic MADD variants. Additionally, our results suggest that patients with MADD variants are likely to have variable degrees of arthrogryposis, structural brain, congenital heart disease, and genital anomalies. The variability of the clinical phenotype of MADD poses challenges to genetic counseling and warrants a comprehensive clinical evaluation and long-term multidisciplinary follow-up of these individuals. Future studies will decipher the precise mechanisms through which MADD deficiency leads to arthrogryposis, genital anomalies and structural brain anomalies.

留言 (0)